Fibrosis is characterized by the excessive accumulation of extra cellular matrix (ECM) components. It is a physiological response to pathological stimuli that helps to confine injuries. However, the prolonged activation of this process results in adverse tissue remodeling, which can ultimately affect the structure and function of organs (adverse remodeling).
- fibrosis
- fibroblast
- TGF-β
- GSK-3
- GRK
- p38
1. Introduction
Fibrosis is characterized by the excessive accumulation of extra cellular matrix (ECM) components. It is a physiological response to pathological stimuli that helps to confine injuries. However, the prolonged activation of this process results in adverse tissue remodeling, which can ultimately affect the structure and function of organs (adverse remodeling). Fibroblasts (FBs) are the major contributor to fibrosis. Previous studies report that the epicardium, endothelial cells, bone-marrow-derived cells, and perivascular cells could be the origins of activated FBs [1][2][3][4][5][6][7][8]. However, recent genetic lineage tracing experiments have confirmed that most activated FBs in the heart originate from tissue-resident FBs [9][10][11][12]. In a normal heart, FBs are quiescent/resting/non-activated. However, in the injured heart, FBs go through a continuum of transient intermediary states and contribute significantly to the cardiac remodeling process [13][14][15][16]. For instance, when myocardial infarction (MI) occurs, a range of stimuli activate resting FBs, giving rise to a new cell state known as activated FBs/myofibroblast. Activated FBs are pro-inflammatory, hyper-secretory, and hyper-migratory in nature. As the healing phase progresses towards scar formation, activated FBs acquire anti-inflammatory and pro-angiogenic phenotypes. They secrete cytokines, ECM components, and other necessary paracrine factors that are required for wound healing. After scar maturation, FBs regress to the quiescent stage or acquire a specialized phenotype (matrifibrocyte) and remain in the matured scar [14][16]. A better understanding of the mechanisms underlying fibroblast activation and fibrosis may provide a novel therapeutic target for managing adverse fibrotic remodeling in the diseased heart.
2. Studies of FB-Specific In Vivo Mouse Models
As noted above, traditionally, most of the research on fibroblast activation and fibrosis was centered on the canonical TGF-β signaling pathway [17][18][19]. However, the recent emergence of these conditional FB-specific mouse models has facilitated the identification of several additional novel pathways that are critical to fibroblast activation and fibrosis. Studies carried out with FB-specific, genetically manipulated mouse models are listed in Table 1. These pathways may operate independently or in co-ordination with TGF-β signaling. On the other hand, these updated tools have helped to redefine several hypotheses and concepts that exclusively relied on isolated culture models or mouse models that were later identified as non-specific. These included the views regarding the sources of FBs in the failing heart and the ability of activated fibroblasts to revert to the quiescent stage. Specifically, in complete disagreement with traditional beliefs, the new genetic models identified that most activated fibroblasts in the failing heart originate from existing resident fibroblasts, and activated fibroblasts can revert to the quiescent stage, once the stress is released [9][14]. These findings have enormous implications for human health; for example, the latter condition is directly comparable to that of patients with ventricular assistance devices (VAD). Furthermore, the discovery of fibroblast’s ability to revert to the quiescent stage opens the door to the potential reverse remodeling of fibrotic hearts.
Table 1. Studies carried out with FB-specific, genetically manipulated mouse models.
| Target Gene | Promoter Used for Cre Expression | Major Findings | References |
|---|---|---|---|
| Tgfbr1/2, Smad2, Smad3 | Postn | FB-specific deletion of Tgfbr1/2 or Smad3, but not Smad2, markedly reduced fibrosis in pressure-overloaded mouse hearts as well as fibrosis mediated by heart-specific, latency-resistant TGF-β mutant transgene. | [20] |
| Smad3 | Postn | In pressure-overloaded hearts, the protective actions of the myofibroblasts were mediated through Smad3-dependent matrix-preserving program | [21] |
| Smad3 | Postn | FB-specific Smad3 loss impaired scar remodeling and increased the incidence of late rupture post-MI | [22] |
| Tgfbr2 | Postn | Tgfbr2 ablation in the myofibroblast prevented fibrosis and cardiac dysfunction in mouse model of cMyBP-C-induced cardiomyopathy | [23] |
| Gsk3b | Postn | FB-specific deletion of GSK-3β lead to the hyperactivation of SMAD-3, resulting in excessive fibrotic remodeling and cardiac dysfunction after myocardial infarction. | [24] |
| Gsk3a | Tcf21 and Postn | In pressure-overloaded hearts, FB-specific GSK-3α mediated pro-fibrotic effects through an ERK-IL-11 circuit that operated independently of TGF-β/SMAD3 signaling | [25] |
| Ctnb1 | Tcf21 and Postn | Loss of β-catenin in fibroblasts attenuated pressure-overload-induced cardiac fibrosis | [26] |
| p38 | Tcf21 and Postn | FB-specific deletion of p38 attenuated myofibroblasts transformation and fibrosis. Conversely, transgenic mice expressing constitutively active p38 in FB specific manner develops fibrosis in multiple organs. | [27] |
| p38 | Postn | Spatial variations in collagen organization regulated cardiac fibroblast phenotype through the mechanical activation of p38-YAP-TEAD signaling | [28] |
| Grk2 | Postn | Ablation of GRK2 in activated fibroblasts significantly reduced myofibroblast transformation and fibrosis and showed cardiovascular protection post-I/R injury | [29] |
| Lats1/2 | Tcf21 | FB-specific deletion of Lats1 and Lats2 initiated a self-perpetuating fibrotic response in the uninjured adult heart that was exacerbated by MI | [30] |
| Yap | Tcf21 | FB-specific deletion of YAP prevented MI-induced cardiac fibrosis and dysfunction through MRTF-A inhibition. | [31] |
| Htr2b | Tcf21 and Postn | Deletion of 5-HT2B receptor signaling in fibroblast prevented border zone expansion and improved microstructural remodeling after MI | [32] |
| Hsp47 | Postn | Myofibroblast-specific ablation of Hsp47 blocked fibrosis in mouse models of pressure overload, MI and, muscular dystrophy | [33] |
| Sox9 | Postn | FB-specific deletion of Sox9 ameliorated MI-induced left ventricular dysfunction, inflammation, and myocardial scarring | [34] |
| Kcnk2 | Tcf21 | FB-specific deletion of TREK1 prevented pressure-overload-induced deterioration in cardiac function | [35] |
| Rock2 | Postn | Deletion of ROCK2 in fibroblast attenuated cardiac hypertrophy, fibrosis, and diastolic dysfunction in mice subjected to chronic Ang-II infusion | [36] |
| Fn1 | Tcf21 | FB-specific fibronectin gene ablation ameliorated adverse cardiac remodeling and fibrosis post I/R | [37] |
| Prkaa1 | Postn | AMPKα1 deletion in myofibroblasts exacerbated post-MI adverse fibrotic remodeling | [38] |
| Sptbn4 | Postn | FB-specific deletion of βIV-spectrin aggravated Ang-II induced fibrosis and cardiac dysfunction. | [39] |
| Pmca4 | Postn | FB-deletion of PMCA4 reduced TAC-induced hypertrophy and cardiac dysfunction | [40] |
| Mbnl1 | Tcf21 and Postn | Deletion of MBNL1 impaired the fibrotic phase of wound healing in mouse models of MI. | [41] |
| Klf5 | Postn | FB–specific KLF5 deletion ameliorated TAC-induced cardiac hypertrophy and fibrosis | [42] |
| Postn | Tcf21 and Postn | Ablation periostin expressing FBs reduced collagen production and scar formation after MI. | [9] |
| Postn | Postn | Ablation of periostin expressing FBs reduced fibrosis and improved cardiac function in mice subjected to chronic Ang-II infusion as well as in mice after MI | [43] |
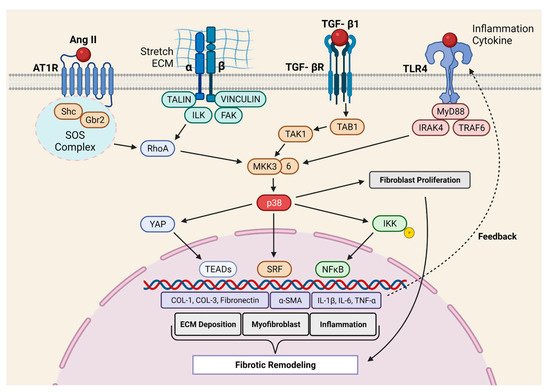
Molkentin et al. provided the first direct evidence for the role of p38 in cardiac fibrosis [27]. They developed genetic mouse models in which p38α could be deleted from fibroblast or myofibroblast using tamoxifen-inducible Tcf21- and Postn- promoter-driven Cre recombinase, respectively. The deletion of p38α from the fibroblast prevented myofibroblast transformation and reduced fibrosis in two different mouse models of cardiac injury (IR and chronic neurohumoral-AngII stimulation). Fibroblast-specific p38α KO mice showed a higher incidence of scar rupture and 100% mortality after MI, thereby highlighting the critical role of this signaling axis in maintaining the structural integrity of the injured myocardium. Conversely, the expression of constitutively active p38α in fibroblast led to the development of cardiac fibrosis in transgenic mice, even in the absence of injury signals, further supporting the crucial role of p38α in fibrosis. In another study, Bageghni et al. [44] deleted p38α from fibroblast using tamoxifen-inducible Col1a2-Cre-ER(T) and observed protection against cardiac hypertrophy induced by isoproterenol (ꞵ-adrenergic receptor agonist). The authors further demonstrated that FB-p38α regulates cardiomyocyte hypertrophy in a paracrine manner through IL-6 secretion. Recently, the Davis group [28] engineered a biomimetic that recapitulates the spatial variations in collagen organization seen in ischemic hearts. Using this novel tool, the authors showed that topological disorganizations in the ECM lead to p38-dependent YAP stabilization in FBs. Indeed, YAP promotes myofibroblast transformation and myocardial fibrosis. Taken together, the p38 MAPK signaling is among the best-characterized positive regulators of fibroblast activation and myocardial fibrosis ( Figure 1 ).
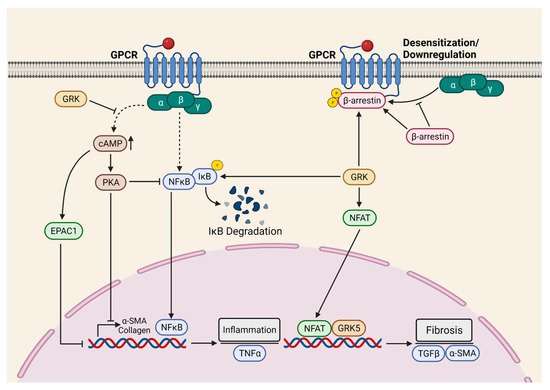
To determine the role of cardiac fibroblast GRK2 in myocardial fibrosis, Koch et al. employed inducible fibroblast-specific GRK2 KOs [45]. Indeed, cardiac fibroblast GRK2 deletion protected against ischemia/reperfusion (I/R)-induced cardiac injury and adverse remodeling [45]. Consistently, pharmacological inhibition or the targeting of GRK2 in activated fibroblast attenuated pathological myofibroblast activation, interstitial fibrosis, and HF progression [29]. These protective effects were associated with a reduction in fibrotic and inflammatory responses in the re-perfused hearts. Mechanistically, it was proposed that GRK2 mediates pro-fibrotic effects by modulating cAMP levels in fibroblasts. Furthermore, GRK2 acts as a positive regulator of NF-κB signaling and promotes inflammatory cytokine secretion in the ischemic heart. The Koch laboratory employed two different mouse models, specifically MI and in vivo AngII infusion, to investigate the fibroblast-specific role of GRK5 in the pathogenesis of cardiac diseases. In both models, the FB-specific deletion of GRK5 prevented adverse cardiac remodeling and improved systolic function. Furthermore, the authors demonstrated that non-canonical interaction between GRK5 and NFAT potentiates NFAT: DNA binding, thereby inducing the transcription of NFAT-mediated fibrotic genes [46]. Additionally, the activation of β2-adrenergic receptors (β2AR) leads to the proliferation of cardiac proliferation and fibrosis through the Gαs/ERK1/2-dependent IL-6 secretion [47]. However, the role of β2AR in cardiac fibrosis needs further validation with conditional FB-specific mouse models in an in vivo setting. Taken together, GPCRs-mediated signaling, specifically β2AR, GRK2, and GRK5, is the critical positive regulator of myocardial fibrosis, therefore representing a novel therapeutic target for the limitation of excessive myofibroblast activation and interstitial fibrosis in the diseased heart ( Figure 2 ).
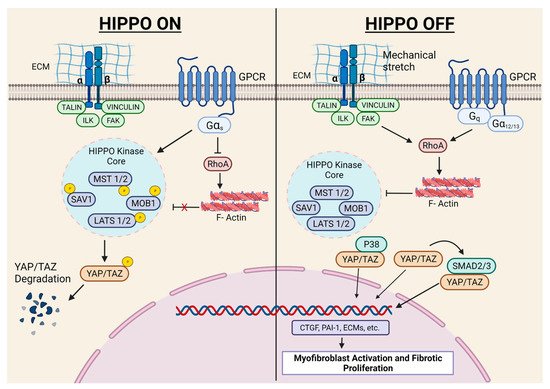
The Hippo pathway plays a critical role in cardiac development, cardiomyocyte biology, and regeneration, which has recently been elegantly reviewed [48]. Herein, we will exclusively focus on in vivo studies with FB-specific mouse models investigating the role of this signaling in fibroblast biology and fibrosis. Fransisco et al. [31] showed that YAP expression increased in FBs after MI, and FB-specific YAP ablation attenuated MI-induced cardiac dysfunction and fibrosis. The authors further demonstrated that YAP promoted myofibroblast differentiation and ECM gene expression through MRTF-A. Martin’s laboratory demonstrated that Hippo signaling promoted epicardial progenitors to fibroblast transition during embryonic development [49]. In another study, the same group conditionally deleted the Hippo pathway kinases LATS1 and LATS2 from adult mouse cardiac fibroblasts. Interestingly, the ablation of LATS1/2 from adult resting cardiac FBs caused spontaneous myofibroblast transformation, cardiac fibrosis, and systolic dysfunction, even in the absence of any cardiac insult. Moreover, the basal fibrotic response (without injury) became more severe in LATS1/2 KOs post-MI, resulting in a poor survival rate. The authors of this study employed single-cell transcriptome analysis and demonstrated that LATS1/2 are essential to the maintenance of FBs in the resting state [30]. These findings are important since there is a general belief that fibroblasts have a minimal role in resting heart physiology. Indeed, most of the research on myocardial fibrosis is limited to diseased conditions. Thus, future investigations are needed to identify the physiological role of fibroblasts in the healthy heart ( Figure 3 ).
3. Conclusions and Future Perspectives
As discussed throughout this review, the recent emergence of the conditional FB-specific mouse model revolutionized the area of cardiac fibroblast biology and myocardial fibrosis research. As a result, the last decade was productive, leading to a paradigm shift towards the idea that fibrosis is not merely a secondary effect of developing pathology and that fibroblasts are not just ECM-producing cells. In fact, numerous studies showed that FB-specific gene targeting can lead to a robust cardiac phenotype, including cardiomyocyte hypertrophy. Indeed, the animals studied demonstrated intact gene expression in all other cells, including the cardiomyocytes. Thus, the last decade saw remarkable progress in our understanding of cardiac fibroblasts’ role in myocardial pathophysiology. There was reasonable success on the mechanism front as well; in addition to the historical profibrotic canonical TGF-β1 pathway, numerous new pro- and anti-fibrotic pathways were identified. However, although substantial progress has been made, it will require a great deal of effort to transform these early bench discoveries into much-needed anti-fibrotic therapies for patients. Specifically, most of the work performed is focused on linear pathways; it is conceivable that these newly identified pathways operate through multiple crosstalk. These signaling circuits and missing links are yet to be established. An upcoming area of enormous potential is the cellular crosstalk among cardiac cells (e.g., fibroblasts to cardiomyocytes) and the circulation (immune cells). The area of fibroblast crosstalk with the immune system and its role in myocardial injury, healing, regeneration, and fibrosis is gaining a lot of interest and is currently under intense investigation by multiple groups. Furthermore, the interplay of fibroblast and inflammation is proposed to play a critical role in the pathogenesis of the comparatively understudied HFpEF syndrome. Regrettably, due to the lack of well-optimized animal models of HFpEF, the progress in this exciting area has been slow and is only just starting to gain some momentum. In the same vein, cardiomyocytes and fibroblast cellular crosstalk has been noted in multiple settings. However, the precise mechanism is not known and is currently under intense investigation by numerous groups. Another emerging area is fibroblast heterogeneity within organs and across various tissues. This line of research with single-cell multi-omics and advanced genomics technologies will be critical to the identification of commonalities and heterogeneity among fibrotic diseases across organs and could play a crucial role in drug repurposing. Indeed, we have recently reported the potential of repurposing Nintedanib (an FDA-approved kinase inhibitor for pulmonary fibrosis) to combat myocardial fibrosis, pathological cardiac remodeling, and dysfunction [50]. The repurposing of authorized anti-fibrotic agents is certainly a “low hanging fruit,” and this line of investigation should be prioritized. Moreover, we anticipate that various fibroblast subpopulations may have a distinct role in the repair and remodeling of the injured heart. Efforts from multiple laboratories with the single-cell RNAseq approach have paved the way towards the identification of the specific markers of different fibroblast subpopulations. We speculate that this knowledge base will help to create future FB-specific mouse models with the ability to target particular FB subpopulations. Finally, it is clear that a better understanding of the profibrotic signaling network may provide a promising new therapeutic target for managing myocardial fibrosis. An effective translation of these new findings will need rigorous verification in human tissues and human tissue-based culture systems, such as pluripotent cell-derived organoids (human tissue chips). There is great optimism that with these newly optimized models and identified pathways, the area of myocardial fibrosis research is set to see another round of growth and productivity. We anticipate that these efforts will enable new approaches to the prevention, treatment, and, hopefully, even the reversal of myocardial fibrosis in diseased hearts.
This entry is adapted from the peer-reviewed paper 10.3390/cells10092412
References
- Kramann, R.; Schneider, R.K.; DiRocco, D.P.; Machado, F.; Fleig, S.; Bondzie, P.A.; Henderson, J.M.; Ebert, B.L.; Humphreys, B.D. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 2015, 16, 51–66.
- Ieronimakis, N.; Hays, A.L.; Janebodin, K.; Mahoney, W.M., Jr.; Duffield, J.S.; Majesky, M.W.; Reyes, M. Coronary adventitial cells are linked to perivascular cardiac fibrosis via TGFbeta1 signaling in the mdx mouse model of Duchenne muscular dystrophy. J. Mol. Cell. Cardiol. 2013, 63, 122–134.
- Van Wijk, B.; Gunst, Q.D.; Moorman, A.F.; van den Hoff, M.J. Cardiac regeneration from activated epicardium. PLoS ONE 2012, 7, e44692.
- Zhou, B.; Honor, L.B.; He, H.; Ma, Q.; Oh, J.H.; Butterfield, C.; Lin, R.Z.; Melero-Martin, J.M.; Dolmatova, E.; Duffy, H.S.; et al. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J. Clin. Investig. 2011, 121, 1894–1904.
- Widyantoro, B.; Emoto, N.; Nakayama, K.; Anggrahini, D.W.; Adiarto, S.; Iwasa, N.; Yagi, K.; Miyagawa, K.; Rikitake, Y.; Suzuki, T.; et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation 2010, 121, 2407–2418.
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961.
- Haudek, S.B.; Xia, Y.; Huebener, P.; Lee, J.M.; Carlson, S.; Crawford, J.R.; Pilling, D.; Gomer, R.H.; Trial, J.; Frangogiannis, N.G.; et al. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 18284–18289.
- Mollmann, H.; Nef, H.M.; Kostin, S.; von Kalle, C.; Pilz, I.; Weber, M.; Schaper, J.; Hamm, C.W.; Elsasser, A. Bone marrow-derived cells contribute to infarct remodelling. Cardiovasc. Res. 2006, 71, 661–671.
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; SC, J.L.; Aronow, B.J.; Tallquist, M.D.; et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 12260.
- Ruiz-Villalba, A.; Simon, A.M.; Pogontke, C.; Castillo, M.I.; Abizanda, G.; Pelacho, B.; Sanchez-Dominguez, R.; Segovia, J.C.; Prosper, F.; Perez-Pomares, J.M. Interacting resident epicardium-derived fibroblasts and recruited bone marrow cells form myocardial infarction scar. J. Am. Coll. Cardiol. 2015, 65, 2057–2066.
- Ali, S.R.; Ranjbarvaziri, S.; Talkhabi, M.; Zhao, P.; Subat, A.; Hojjat, A.; Kamran, P.; Muller, A.M.; Volz, K.S.; Tang, Z.; et al. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ. Res. 2014, 115, 625–635.
- Moore-Morris, T.; Guimaraes-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934.
- Tallquist, M.D. Cardiac Fibroblast Diversity. Annu. Rev. Physiol. 2020, 82, 63–78.
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 2018, 128, 2127–2143.
- Tallquist, M.D.; Molkentin, J.D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 2017, 14, 484–491.
- Ma, Y.; Iyer, R.P.; Jung, M.; Czubryt, M.P.; Lindsey, M.L. Cardiac Fibroblast Activation Post-Myocardial Infarction: Current Knowledge Gaps. Trends Pharmacol. Sci. 2017, 38, 448–458.
- Frangogiannis, N. Transforming growth factor-beta in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103.
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338.
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-beta signaling in fibrosis. Growth Factors 2011, 29, 196–202.
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.J.; et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783.
- Russo, I.; Cavalera, M.; Huang, S.; Su, Y.; Hanna, A.; Chen, B.; Shinde, A.V.; Conway, S.J.; Graff, J.; Frangogiannis, N.G. Protective Effects of Activated Myofibroblasts in the Pressure-Overloaded Myocardium Are Mediated Through Smad-Dependent Activation of a Matrix-Preserving Program. Circ. Res. 2019, 124, 1214–1227.
- Kong, P.; Shinde, A.V.; Su, Y.; Russo, I.; Chen, B.; Saxena, A.; Conway, S.J.; Graff, J.M.; Frangogiannis, N.G. Opposing Actions of Fibroblast and Cardiomyocyte Smad3 Signaling in the Infarcted Myocardium. Circulation 2018, 137, 707–724.
- Meng, Q.; Bhandary, B.; Bhuiyan, M.S.; James, J.; Osinska, H.; Valiente-Alandi, I.; Shay-Winkler, K.; Gulick, J.; Molkentin, J.D.; Blaxall, B.C.; et al. Myofibroblast-Specific TGFbeta Receptor II Signaling in the Fibrotic Response to Cardiac Myosin Binding Protein C-Induced Cardiomyopathy. Circ. Res. 2018, 123, 1285–1297.
- Lal, H.; Ahmad, F.; Zhou, J.; Yu, J.E.; Vagnozzi, R.J.; Guo, Y.; Yu, D.; Tsai, E.J.; Woodgett, J.; Gao, E.; et al. Cardiac fibroblast glycogen synthase kinase-3beta regulates ventricular remodeling and dysfunction in ischemic heart. Circulation 2014, 130, 419–430.
- Umbarkar, P.; Tousif, S.; Singh, A.P.; Anderson, J.C.; Zhang, Q.; Lal, H. Cardiac fibroblast GSK-3α mediates adverse myocardial fibrosis via IL-11 and ERK pathway. bioRxiv 2021.
- Xiang, F.L.; Fang, M.; Yutzey, K.E. Loss of beta-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat. Commun. 2017, 8, 712.
- Molkentin, J.D.; Bugg, D.; Ghearing, N.; Dorn, L.E.; Kim, P.; Sargent, M.A.; Gunaje, J.; Otsu, K.; Davis, J. Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation 2017, 136, 549–561.
- Bugg, D.; Bretherton, R.; Kim, P.; Olszewski, E.; Nagle, A.; Schumacher, A.E.; Chu, N.; Gunaje, J.; DeForest, C.A.; Stevens, K.; et al. Infarct Collagen Topography Regulates Fibroblast Fate via p38-Yes-Associated Protein Transcriptional Enhanced Associate Domain Signals. Circ. Res. 2020, 127, 1306–1322.
- Travers, J.G.; Kamal, F.A.; Valiente-Alandi, I.; Nieman, M.L.; Sargent, M.A.; Lorenz, J.N.; Molkentin, J.D.; Blaxall, B.C. Pharmacological and Activated Fibroblast Targeting of Gbetagamma-GRK2 After Myocardial Ischemia Attenuates Heart Failure Progression. J. Am. Coll. Cardiol. 2017, 70, 958–971.
- Xiao, Y.; Hill, M.C.; Li, L.; Deshmukh, V.; Martin, T.J.; Wang, J.; Martin, J.F. Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes. Dev. 2019, 33, 1491–1505.
- Francisco, J.; Zhang, Y.; Jeong, J.I.; Mizushima, W.; Ikeda, S.; Ivessa, A.; Oka, S.; Zhai, P.; Tallquist, M.D.; Del Re, D.P. Blockade of Fibroblast YAP Attenuates Cardiac Fibrosis and Dysfunction Through MRTF-A Inhibition. JACC Basic Transl. Sci. 2020, 5, 931–945.
- Snider, J.C.; Riley, L.A.; Mallory, N.T.; Bersi, M.R.; Umbarkar, P.; Gautam, R.; Zhang, Q.; Mahadevan-Jansen, A.; Hatzopoulos, A.K.; Maroteaux, L.; et al. Targeting 5-HT2B Receptor Signaling Prevents Border Zone Expansion and Improves Microstructural Remodeling After Myocardial Infarction. Circulation 2021, 143, 1317–1330.
- Khalil, H.; Kanisicak, O.; Vagnozzi, R.J.; Johansen, A.K.; Maliken, B.D.; Prasad, V.; Boyer, J.G.; Brody, M.J.; Schips, T.; Kilian, K.K.; et al. Cell-specific ablation of Hsp47 defines the collagen-producing cells in the injured heart. JCI Insight 2019, 4, e128722.
- Scharf, G.M.; Kilian, K.; Cordero, J.; Wang, Y.; Grund, A.; Hofmann, M.; Froese, N.; Wang, X.; Kispert, A.; Kist, R.; et al. Inactivation of Sox9 in fibroblasts reduces cardiac fibrosis and inflammation. JCI Insight 2019, 5, e126721.
- Abraham, D.M.; Lee, T.E.; Watson, L.J.; Mao, L.; Chandok, G.; Wang, H.G.; Frangakis, S.; Pitt, G.S.; Shah, S.H.; Wolf, M.J.; et al. The two-pore domain potassium channel TREK-1 mediates cardiac fibrosis and diastolic dysfunction. J. Clin. Investig. 2018, 128, 4843–4855.
- Shimizu, T.; Narang, N.; Chen, P.; Yu, B.; Knapp, M.; Janardanan, J.; Blair, J.; Liao, J.K. Fibroblast deletion of ROCK2 attenuates cardiac hypertrophy, fibrosis, and diastolic dysfunction. JCI Insight 2017, 2, e93187.
- Valiente-Alandi, I.; Potter, S.J.; Salvador, A.M.; Schafer, A.E.; Schips, T.; Carrillo-Salinas, F.; Gibson, A.M.; Nieman, M.L.; Perkins, C.; Sargent, M.A.; et al. Inhibiting Fibronectin Attenuates Fibrosis and Improves Cardiac Function in a Model of Heart Failure. Circulation 2018, 138, 1236–1252.
- Dufeys, C.; Daskalopoulos, E.P.; Castanares-Zapatero, D.; Conway, S.J.; Ginion, A.; Bouzin, C.; Ambroise, J.; Bearzatto, B.; Gala, J.L.; Heymans, S.; et al. AMPKalpha1 deletion in myofibroblasts exacerbates post-myocardial infarction fibrosis by a connexin 43 mechanism. Basic Res. Cardiol. 2021, 116, 10.
- Patel, N.J.; Nassal, D.M.; Greer-Short, A.D.; Unudurthi, S.D.; Scandling, B.W.; Gratz, D.; Xu, X.; Kalyanasundaram, A.; Fedorov, V.V.; Accornero, F.; et al. betaIV-Spectrin/STAT3 complex regulates fibroblast phenotype, fibrosis, and cardiac function. JCI Insight 2019, 4, e131046.
- Mohamed, T.M.A.; Abou-Leisa, R.; Stafford, N.; Maqsood, A.; Zi, M.; Prehar, S.; Baudoin-Stanley, F.; Wang, X.; Neyses, L.; Cartwright, E.J.; et al. The plasma membrane calcium ATPase 4 signalling in cardiac fibroblasts mediates cardiomyocyte hypertrophy. Nat. Commun. 2016, 7, 11074.
- Davis, J.; Salomonis, N.; Ghearing, N.; Lin, S.C.; Kwong, J.Q.; Mohan, A.; Swanson, M.S.; Molkentin, J.D. MBNL1-mediated regulation of differentiation RNAs promotes myofibroblast transformation and the fibrotic response. Nat. Commun. 2015, 6, 10084.
- Takeda, N.; Manabe, I.; Uchino, Y.; Eguchi, K.; Matsumoto, S.; Nishimura, S.; Shindo, T.; Sano, M.; Otsu, K.; Snider, P.; et al. Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. J. Clin. Investig. 2010, 120, 254–265.
- Kaur, H.; Takefuji, M.; Ngai, C.Y.; Carvalho, J.; Bayer, J.; Wietelmann, A.; Poetsch, A.; Hoelper, S.; Conway, S.J.; Mollmann, H.; et al. Targeted Ablation of Periostin-Expressing Activated Fibroblasts Prevents Adverse Cardiac Remodeling in Mice. Circ. Res. 2016, 118, 1906–1917.
- Bageghni, S.A.; Hemmings, K.E.; Zava, N.; Denton, C.P.; Porter, K.E.; Ainscough, J.F.X.; Drinkhill, M.J.; Turner, N.A. Cardiac fibroblast-specific p38alpha MAP kinase promotes cardiac hypertrophy via a putative paracrine interleukin-6 signaling mechanism. FASEB J. 2018, 32, 4941–4954.
- Woodall, M.C.; Woodall, B.P.; Gao, E.; Yuan, A.; Koch, W.J. Cardiac Fibroblast GRK2 Deletion Enhances Contractility and Remodeling Following Ischemia/Reperfusion Injury. Circ. Res. 2016, 119, 1116–1127.
- Eguchi, A.; Coleman, R.; Gresham, K.; Gao, E.; Ibetti, J.; Chuprun, J.K.; Koch, W.J. GRK5 is a regulator of fibroblast activation and cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2012854118.
- Tanner, M.A.; Thomas, T.P.; Maitz, C.A.; Grisanti, L.A. beta2-Adrenergic Receptors Increase Cardiac Fibroblast Proliferation Through the Galphas/ERK1/2-Dependent Secretion of Interleukin-6. Int. J. Mol. Sci. 2020, 21, 8507.
- Wang, J.; Liu, S.; Heallen, T.; Martin, J.F. The Hippo pathway in the heart: Pivotal roles in development, disease, and regeneration. Nat. Rev. Cardiol. 2018, 15, 672–684.
- Xiao, Y.; Hill, M.C.; Zhang, M.; Martin, T.J.; Morikawa, Y.; Wang, S.; Moise, A.R.; Wythe, J.D.; Martin, J.F. Hippo Signaling Plays an Essential Role in Cell State Transitions during Cardiac Fibroblast Development. Dev. Cell 2018, 45, 153–169.
- Umbarkar, P.; Singh, A.P.; Tousif, S.; Zhang, Q.; Sethu, P.; Lal, H. Repurposing Nintedanib for pathological cardiac remodeling and dysfunction. Pharmacol. Res. 2021, 169, 105605.