Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Protein tyrosine kinases, especially receptor tyrosine kinases, have dominated the cancer therapeutics sphere as proteins that can be inhibited to selectively target cancer. However, protein tyrosine phosphatases (PTPs) are also an emerging target. Though historically known as negative regulators of the oncogenic tyrosine kinases, PTPs are now known to be both tumor-suppressive and oncogenic.
- breast cancer
- gastric cancer
- prostate cancer
- PTP
- phosphatase
- receptor tyrosine kinase
- protein tyrosine phosphatase
- RTK
1. Introduction
Protein tyrosine phosphatases (PTPs) function to remove phosphate groups from proteins. Historically, PTPs have been thought of as being opposite to protein tyrosine kinases (PTKs), since PTKs are enzymes that add phosphate groups to proteins. One major focus of cancer research involves understanding how a subset of PTKs, known as receptor tyrosine kinases (RTKs), enables the progression and survival of cancer. RTKs, many of which are growth factor receptors, have intracellular domains that auto-phosphorylate upon ligand binding [1][2]. This induces the activation of kinases that phosphorylate downstream proteins, which can further phosphorylate even more downstream proteins [1][2]. The phosphorylation cascade results in the activation of many normal cell signaling pathways that induce proliferation, differentiation, survival, and cell migration [1][2]. Some important growth-promoting signaling proteins, which are recruited to the activated RTK’s intracellular phosphotyrosine residues, include phosphoinositide 3 kinase (PI3K), Ras, and Janus kinase (JAK) [1][2][3]. PI3K, Ras, and JAK generate the PI3K-AKT, mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK), and JAK-STAT signaling cascades, respectively [1][2][3]. Therefore, upregulation of RTK expression is a key molecular mechanism that underlies many cancers. On the other hand, PTPs were thought to inhibit the actions of RTKs by performing the reciprocal action of kinases, which is to dephosphorylate proteins [4][5]. However, it is now known that in normal physiology, some PTP dephosphorylation balances RTK and PTK phosphorylation to regulate cell signaling dynamics, while other PTP subtypes are known to be proto-oncogenic by enhancing growth factor and survival signaling pathways [4][5].
Protein tyrosine phosphatases can be classified as tyrosine-specific or dual-specific [4][5][6]. Dual-specific PTPs are able to dephosphorylate both tyrosine and serine/threonine residues [4][5][6]. Furthermore, PTPs can also be grouped based on their location within the cell. PTPs that span the cell membrane are receptor types, while PTPs within the cytosol are non-receptor types [4][5][6]. Despite these classifications, PTPs exhibit oncogenic or tumor-suppressive activity via their ability to dephosphorylate proteins [4][5]. It is the combination of PTP and the unique pathway it operates on that determines whether dephosphorylation will lead to promotion or hindrance of tumorigenesis. Hence, a given PTP can have the same function or different function across many organs. A PTP that exhibits loss of function in one cancer can have a gain of function mutation in a different cancer.
Some heavily studied PTPs can provide insight into the general mechanisms of tumor-suppressive and oncogenic PTPs. Src homology-2-containing protein tyrosine phosphatase 2 (SHP-2) is a non-receptor, tyrosine-specific, PTP encoded from the PTPN11 gene that binds to the RTK intracellular domain via phosphotyrosine residues [7][8][9]. The Src homology 2 domain of SHP-2, once occupied by tyrosine residues, no longer impedes the phosphatase activity of SHP-2 [7][9][10]. SHP-2 dephosphorylates certain proteins or residues that suppress the function of Ras inhibitors and Src family kinase (SFK) inhibitors, thus enhancing the activity of these tumorigenic pathways [11][12][13][14]. In other words, SHP-2, through its ability to dephosphorylate and inactivate SFK and Ras inhibitors, can promote the PI3K and MAPK/ERK signaling that arises downstream of activated RTKs [11][12][13][14][15][16][17][18]. SHP-2 is an example of a PTP that seems to exhibit oncogenic activity in many cancer cell types, which will be covered later in the review.
Phosphatase of regenerating liver 3 (PRL-3), also known as PTP4A3, is a dual-specific, non-receptor PTP that is highly expressed in many tumors [6]. Upon activation of growth factor receptors like platelet-derived growth factor (PDGF), Src, a tyrosine kinase that is a part of the Src family of kinases, phosphorylates and activates PRL-3 at specific amino acid residues [19]. Once activated, PRL-3 can act upstream of Src kinase by downregulating expression of Src kinase inhibitor c-Srk kinase (Csk), thereby promoting the oncogenic RAS-RAF-MEK-ERK pathway downstream of Src kinase [20]. PRL-3, like SHP-2, also exhibits tumor-promoting activity across different cancer types.
In contrast, protein tyrosine phosphatase 1b (PTP1b), a non-receptor, tyrosine-specific PTP, has been implicated in both hindering and promoting cancer [6]. In studies that were mainly done in fibroblasts and endothelial cells (cell types that can enhance tumorigenesis), inhibition of PTP1b proved to enhance migration, while overexpression of PTP1b suppressed migration [21][22]. One of the major molecular mechanisms underlying PTP1b’s ability to reduce migration is the dephosphorylation and inactivation of p130cas, which impedes the scaffold protein’s ability to recruit and activate Rac [23][24][25]. The Rac signaling pathway is crucial in promoting actin polymerization and lamellipodia formation during cell migration [26]. Rac signaling can also activate matrix metalloproteinases that can degrade the extracellular matrix to promote cancer invasion [23][27]. Thus, the tumor suppressor function of PTP1b is highlighted through its ability to dephosphorylate key proline-rich residues of p130cas and inhibit p130cas-Rac induced migration and invasion of fibroblasts and endothelial cells [25].
2. PTPs That Regulate the JAK-STAT Pathway
Emerging research suggests that PTPs play a key role in regulating different aspects of the JAK-STAT pathway. SHP-1, a non-receptor, tyrosine-specific PTP that exhibits tumor suppressor activity in gastric cancer, was found to dephosphorylate and inhibit STAT3 signaling in vitro and in vivo [28]. Upon increasing SHP-1 expression, cell proliferation was inhibited in vitro, and tumor growth was inhibited in vivo [28]. Additional experiments led researchers to discover that when SHP-1 protein expression was increased, gastric cancer cells exhibited reduced phosphorylation of STAT3, which resulted in downstream inhibition of Cyclin D1 and XIAP (Figure 1) [28]. Furthermore, another study identified that SHP-1 gene expression is reduced in gastric cancer cells owing to promoter region hypermethylation [29]. Overexpression of SHP-1 in gastric cancer cells downregulated the JAK-STAT pathway (decreased phosphorylation of JAK2 and STAT3 due to its dephosphorylation activity) and led to the downregulation of proteins that are important for cell cycle progression, invasion, and angiogenesis (Cyclin D1, MMP-9, VEGF1) (Figure 1) [29]. Both studies identified key molecular mechanisms underlying SHP-1′s ability to inhibit cancer progression, which provides strong evidence that SHP-1 is a tumor suppressor (Table 1). Further research is needed to identify SHP-1 loss of function in gastric cancer as a predictor of poor prognosis and decreased patient survival.
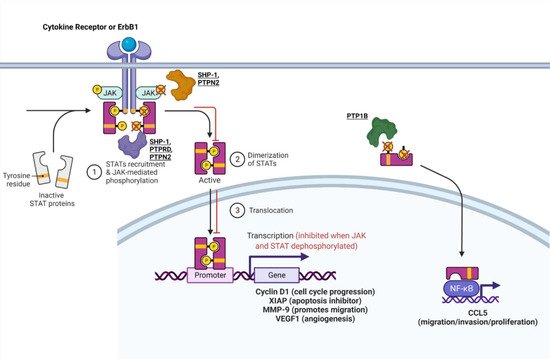
Figure 1. PTPs that regulate the JAK-STAT pathway. The mechanism of some PTPs influence the JAK-STAT or STAT-CCL5 pathway. For a more in-depth discussion of the mechanism of action for each PTP, refer to Table 1. Adapted from “Cytokine Signaling through the JAK-STAT Pathway”, by BioRender.com (accessed on 28 September 2021). Retrieved from https://app.biorender.com/biorender-templates (accessed on 28 September 2021).
Table 1. PTPs that regulate the JAK-STAT pathway.
| PTP | Classification | Cellular/Molecular Function | Oncogene (O)/Tumor Suppressor (TS) | Figure Illustration |
|---|---|---|---|---|
| Src homology region 2 domain containing phosphatase 1 (SHP-1)/Tyrosine protein phosphatase non-receptor type 6 (PTPN6) | Non-receptor, tyrosine-specific | Downregulation of JAK-STAT, XIAP, Cyclin D1, MMP-9, VEGF1 | TS in gastric cancer | Figure 1 |
| Tyrosine protein phosphatase non-receptor type 1 (PTPN1)/Protein Tyrosine Phosphatase 1B (PTP1B) | Non-receptor, tyrosine-specific | Dephosphorylates STAT3, increases CCL5; | O in breast cancer | Figure 1 |
| Tyrosine protein phosphatase non-receptor type 2 (PTPN2) | Non-receptor, tyrosine-specific | Dephosphorylates ErbB1 (HER1), p-JAK, p-STAT | TS in breast cancer | Figure 1 |
| Protein Tyrosine Phosphatase Receptor Type D (PTPRD) | Receptor-type, tyrosine-specific | Dephosphorylates STAT3 | TS in gastric cancer | Figure 1 |
Protein tyrosine phosphatase 1b (PTP1b) promotes tumorigenesis in many types of cancer including breast cancer. It was shown to be overexpressed primarily in HER2+ breast cancers and influence the JAK-STAT pathway [30]. In one study, PTP1b increased tumor size and lymph node metastasis by dephosphorylating phospho-STAT 3, thus increasing CCL5 expression, which is involved in increased cell migration and proliferation (Figure 1) [30]. This finding contradicts the known function of the STAT3 pathway, in which dephosphorylated STAT3 is inactive and typically inhibits tumorigenic pathways. However, the researchers in this study confirmed their findings through the knockdown of PTP1b, which increased phosphorylation of STAT3, resulting in decreased CCL5 expression and diminished cell proliferation, migration, and invasion in MCF-7 cells [30]. Another study confirmed the tumor-promoting function of PTP1b by identifying that homozygous knockdown of PTP1b (PTP1b−/−) was capable of significantly decreasing or delaying tumor formation, while heterozygous knockdown of PTP1b did not have a significant effect [31]. Hence, PTP1b has been proven to enhance the formation, spread, and aggressiveness of breast cancer (Table 1).
Protein tyrosine phosphatase non-receptor type 2 (PTPN2), also known as T-cell PTP, is another tyrosine-specific PTP that may modulate ER+ breast cancer sensitivity to tamoxifen treatment by lowering the JAK-STAT cancerous signaling pathway [32]. Scientists believe that crosstalk between ER and growth factor signaling pathways plays a role in breast cancer’s resistance to treatment [32][33][34]. Since PTPN2 has been shown to inhibit growth through the PI3K/AKT pathway, it may affect this crosstalk [35][36]. In one study, decreased expression of PTPN2 in ER+ breast cancer was associated with increased expression of nuclear p-AKT, resulting in an overall poorer response to tamoxifen treatment in breast cancer [32]. One study provided a possible mechanism for PTPN2 in breast cancer expressing epidermal growth factor receptors (EGFRs) and ER. ErbB1 (EGFR/HER1) along with downstream Janus kinases and STAT3 are substrates for PTPN2 [35][37][38][39][40][41][42]. Therefore, PTPN2 may directly dephosphorylate ERbB1, leading to inhibition of downstream phospho-JAK and phospho-STAT3 [3][37]. PTPN2 may possibly dephosphorylate JAK and STAT3 directly as well (Figure 1), which could reduce phosphorylation of AKT as a result of JAK-STAT-AKT crosstalk [37][43]. Thus, PTPN2 may serve as a marker that predicts poor patient survival overall and poor response to antihormone treatment when it exhibits loss of function in breast cancer (Table 1).
PTP delta (PTPRD) is a tumor-suppressive, tyrosine-specific, PTP in gastric cancer. Patient data from one study showed that reduced PTPRD expression was associated with diminished survival [44]. Additionally, knockdown of PTPRD induced phosphorylation of STAT3, which heightened survival, proliferation, migration, and invasion as confirmed by phenotypic assays (Figure 1) [44].
SHP-1, PTP1b, PTPN2, and PTPRD are all phosphatases that influence the JAK-STAT pathway in gastric and breast cancer. The mechanisms of these phosphatases must be further studied in other cancers to confirm whether their role is identical in other cell types. For example, SHP-1 and PTP1b impact other pathways and will be discussed in later sections.
3. PTPs That Impact SFKs and PTEN
PTP1b, which influences JAK-STAT signaling to induce oncogenesis in breast cancer, can also promote breast cancer by dephosphorylating the inhibitory Tyr527 phosphorylation site and activating Src and downstream signaling pathways, which enhances ERbB2 receptor signaling (Figure 2B) [17][18][45]. In this study, ErbB2 was activated with simultaneous knockdown of PTP1b to see how properties such as apoptosis and cell proliferation were affected [45]. They observed that despite ErbB2 activation, the knockdown of PTP1b resulted in increased apoptosis and decreased cell proliferation [45]. In addition to tumorigenesis and proliferation, PTP1b is also crucial for lymph node metastasis, migration, and invasion, as shown by a study that knocked down PTP1b, resulting in reduced migration and invasion [46]. PTP1b modulated these phenotypes by downregulating PTEN expression, thereby upregulating the AKT pathway and increasing the expression of invasion-promoting proteases MMP-2 and MMP-7 (Figure 2A) [46].
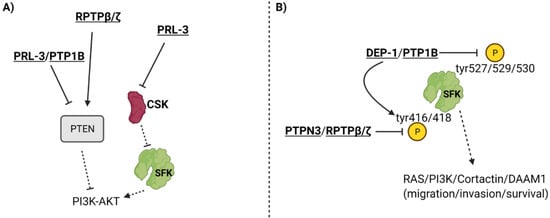
Figure 2. PTPs that impact SFKs and PTEN. PTPs can influence PTEN, CSK (Src Kinase inhibitor) (Panel A), and phosphorylation of the Tyr527/529/530 inhibitory residues and the Tyr416/418 catalytic residues of the Src family of kinases (Panel B). To see how PTEN and SFK function in signaling cascades, refer to Figure 3. For a more in-depth discussion of the mechanism of action for each PTP, refer to Table 2.
Table 2. PTPs that impact SFKs and PTEN to influence PI3K-AKT, Cortactin, and DAAM1 dynamics.
| PTP | Classification | Cellular/Molecular Function | Oncogene (O)/Tumor Suppressor (TS) | Figure Illustration |
|---|---|---|---|---|
| Tyrosine protein phosphatase non-receptor type 1 (PTPN1)/Protein Tyrosine Phosphatase 1B (PTP1B) | Non-receptor, tyrosine-specific | Dephosphorylates Tyr527 residue of Src (activation); Inhibits PTEN expression | O in breast cancer | Figure 2 |
| PTPN1/PTP1B | Non-receptor, tyrosine-specific | Exact mechanism is unknown. May dephosphorylate STAT3, increases CCL5; Dephosphorylates Tyr527 residue of Src (activation); Inhibits PTEN expression | O in prostate cancer | Figure 1 and Figure 2 |
| Phosphatase of Regenerating Liver 3 (PRL-3)/Protein Tyrosine Phosphatase 4A3 (PTP4A3) | Non-receptor, tyrosine-specific | Inhibits PTEN expression, which heightens PI3K-AKT signaling | O in gastric cancer | Figure 2 |
| PRL-3/PTP4A3 | Non-receptor, tyrosine-specific | Not well understood. May heighten Src activity; Inhibits PTEN expression, which heightens PI3K-AKT signaling | O in breast cancer | Figure 2 |
| Receptor protein tyrosine phosphatase beta/zeta (RPTPβ/ζ) | Receptor-type, tyrosine-specific | Reduces Tyr416 phosphorylation of Src and inactivates it; Reduces phosphorylation of and activates PTEN | TS in prostate cancer | Figure 2 |
| Receptor-type tyrosine protein phosphatase eta (PTPRJ)/Density Enhanced Phosphatase 1 (DEP-1) | Receptor-type, tyrosine-specific | Dephosphorylates Src at Tyr529, which increases Src Tyr418 and subsequent Cortactin phosphorylation | O in breast cancer | Figure 2 |
| Protein Tyrosine Phosphatase H1 (PTPH1)/Tyrosine protein phosphatase non-receptor type 3 (PTPN3) | Non-receptor, tyrosine-specific | Dephosphorylates and inhibits Src mediated DAAM1 phosphorylation; Directly inhibits DAAM1 phosphorylation | TS in gastric cancer | Figure 2 |
PTP1b also exhibits pro-cancerous activity in prostate cancer. In prostate cancer cells, neuroendocrine (NE) cells make up a portion of the tumor and can secrete neuropeptides, growth factors, and other hormones [47][48]. Various studies have proven that hormonal therapy leads to NE differentiation of prostate cancer cells, which can lead to higher metastatic potential [47][49][50][51][52][53]. One study revealed that elevated expression levels of PTP1B can lead to NE differentiation in LNCaP cells [47]. In NE-differentiated LNCaP cells, PTP1B is highly expressed and is exclusively localized to the NE-differentiated cells of the prostate cancer [47]. Further research on PTP1b shows that PTP1b knockdown abrogates migration, invasion, and growth in vitro and in vivo, which highlights its importance as a promoter of prostate cancer [54]. Overall, although the exact mechanism of PTP1B is unknown in prostate cancer, it can be concluded that PTP1B plays an oncogenic role in prostate cancer. Its mechanism may be similar to how it functions in breast cancer, but further research is necessary to uncover how PTP1b elicits pro-cancerous activity in prostate cancer (Table 2). PTP1b is a versatile phosphatase that influences many pathways in breast cancer and possibly prostate cancer to promote tumorigenesis; hence, it must be explored more as a cancer-associated phosphatase that can be used for targeted therapy.
Phosphatase of regenerating liver 3 (PRL-3), also known as PTP4A3, is a dual-specific, non-receptor PTP that exhibits tumorigenic activity in gastric cancer via PTEN-dependent interactions that have been identified in many studies. PRL-3 overexpressing gastric cancer cells exhibited increased p-AKT and downstream matrix metalloproteinase (MMP) expression, which was further confirmed by increased migration and invasion in comparison to control cells [55]. This signaling pathway, along with migration and invasion, was inhibited in PRL-3 overexpressing gastric cancer cells treated with PI3K inhibitor, confirming that PRL-3 acts upstream of the PI3K-AKT pathway to induce its activation [55]. Previous studies strengthened this finding by discovering that PRL-3 inhibits phosphatase and tensin homologue (PTEN) expression, a phosphatase that converts the AKT activating phosphatidylinositol (3,4,5)-triphosphate (PIP3) to phosphatidylinositol (4,5)-bisphosphate (PIP2) [56][57]. Therefore, it is possible that PRL-3 acts upstream of PI3K-AKT by inhibiting PTEN, thus promoting PI3K’s ability to generate AKT activating PIP3 (Figure 2A). It is evident that PRL-3 heightens cancer dynamics (Table 2), validating the need for further research into the inhibition of PRL-3 as a specific therapy in gastric cancer.
PRL-3 is a crucial PTP in breast cancer as well. The PTP4A3 gene is overexpressed in 29% of all basal-like breast cancers and may be a prognostic indicator for poor breast cancer patient survival [58]. A study revealed that PRL-3 elicited the growth, survival, and metastatic progression of triple negative breast cancer in vivo and in vitro [58]. Furthermore, PRL-3 knockdown resulted in G1 cell cycle arrest and apoptosis, which identifies PTP4A3 as being necessary for progression of TNBC [58]. PRL-3 also caused cell cycle arrest in estrogen receptor (ER)-positive breast cancer cell lines, indicating the need for more experiments to confirm PRL-3 as a modulator of cellular dynamics in ER+ breast cancer [58]. The mechanism of PRL-3 has not been thoroughly investigated in breast cancer. However, previous studies probing into the mechanism of PRL-3 in other cancers demonstrated that the phosphatase downregulated the expression of another phosphatase known as phosphatase and tensin homologue (PTEN), leading to the upregulation of the PI3K-AKT pathway through signaling modalities that were previously described (Figure 2A) [56][57]. PRL-3 may also act upstream of Src kinase by downregulating the expression of Src kinase inhibitor c-Srk kinase (Csk), which would lead to the upregulation of many oncogenic pathways downstream of Src (Figure 2A) [20]. Overall, strong evidence suggests that PRL-3 has a tumor-promoting role in breast cancer and induces epithelial-to-mesenchymal transition (EMT), growth, and survival (Table 2).
Receptor protein tyrosine phosphatase beta/zeta (RPTPβ/ζ) is a receptor-type, tyrosine-specific PTP that exhibits tumor suppressor activity in prostate cancer through its interaction with Src and PTEN. A study identified RPTPβ/ζ as an inhibitor of tumorigenesis via its ability to reduce phosphorylation of Src catalytic domain Tyr416 residue and inactivate Src mediated pathways (Figure 2) [59]. Additionally, RPTPβ/ζ reduced phosphorylation of and activated PTEN, which is known to reduce PI3K-AKT signaling in addition to downstream MAPK/ERK signaling (Figure 2) [59]. Receptor protein tyrosine phosphatases (RPTPβ/ζ) and syndecan-3 are pleiotrophin-binding transmembrane receptors [60][61][62]. Experiments revealed that pleiotrophin (PTN), despite being known as a growth factor, inhibits EMT of prostate cancer cells by eliciting PTEN activation and Src inactivation through RPTPβ/ζ [59]. However, PTN promotes EMT and migration by phosphorylating and activating Src and downstream oncogenic signals via syndecan-3 receptor in RPTPβ/ζ knockdown cell lines [59]. The study revealed that RPTPβ/ζ may function as a tumor suppressor by inhibiting tumorigenic signaling pathways and attenuating syndecan-3 signaling (Table 2) [59]. This study focused mainly on changes in Src, PTEN, ERK, and migration. Further studies are required to understand how RPTPβ/ζ affects more key players in the PI3K-AKT and MAPK/ERK signaling pathways in addition to whether RPTPβ/ζ influences apoptosis and cell cycle progression through these pathways [63].
DEP-1/PTPRJ is a tyrosine-specific receptor tyrosine phosphatase that is primarily known for its role as a tumor suppressor [64][65]. Yet, one study found that DEP-1 is expressed more in highly aggressive breast cancer types and may contribute to invasion and migration [66]. Researchers found that when DEP-1 was expressed at intermediate levels, it effectively dephosphorylated Src at Tyr529 inhibitory residue and activated the Src kinase pathway, causing the downstream phosphorylation of Cortactin to promote motile, invasive, and metastatic phenotypes [66]. DEP-1 knockdown resulted in the inhibition of the Src/Cortactin pathway, as represented by reduced phosphorylation of these proteins (Src at Tyr418 catalytic residue) (Figure 2B) [66]. To corroborate these molecular results, the study also revealed that DEP-1 knockdown reduced migration, invasion, and secreted MMP-9 [66]. Though DEP-1 seems to promote cancer progression, scientists discovered through transfection experiments at different DNA concentrations that DEP-1 was most effective at moderate levels, whereas extremely increased or decreased expression of DEP-1 resulted in less Src/Cortactin activation [66]. The same study also highlighted that moderate levels of DEP-1 in breast cancer patients were associated with the highest rates of mortality and cancer relapse, compared to lower and higher expression of DEP-1 in patients [66]. Overall, this study showed that DEP-1, though known for its tumor suppressor role, functions via the Src/Cortactin pathway to enhance metastatic properties in aggressive breast cancer cell types and is linked to poorer prognosis in patients (Table 2). DEP-1 may be a PTP that exhibits varying activity across cell types, though further studies are required to confirm this.
PTPN3, also known as PTPH1, is a non-receptor, tyrosine-specific tumor suppressor PTP in gastric cancer. The mechanism of PTPN3 has been identified in previous research. One study identified that knocking down PTPN3 enhanced production of VEGFA in gastric cancer cells [67]. Incubating human umbilical vein endothelial cells (HUVECs) in conditioned media from PTPN3 KD gastric cancer cells resulted in elevated cell viability, migration, invasion, and more robust tube formation of HUVECs [67]. Another potential mechanism underlying PTPN3 activity may be explained by a study performed on lung cancer cells. PTPN3 was found to dephosphorylate Src at catalytic domain Tyr416 residue and inactivate it, leading to reduced phosphorylation and increased inactivation of disheveled associated activator of morphogenesis 1 (DAAM1). DAAM1 is a key protein in regulating actin dynamics, and inactivation of this protein led to reduced cell migration, invasion, and focal adhesion assembly (Figure 2B) [68][69]. In gastric cancer cells, PTPN3 loss has proven to heighten angiogenesis and tumor metastasis (Table 2). Overall, PTP1b, PRL-3, RPTPβ/ζ, DEP-1, and PTPN3 all modulate SFKs or PTEN in different cancers. PTP1b promotes breast cancer through modulation of the JAK-STAT pathway, Src, and PTEN, while PRL-3 expression augments both breast and gastric cancer.
This entry is adapted from the peer-reviewed paper 10.3390/ijms222312865
References
- Regad, T. Targeting RTK Signaling Pathways in Cancer. Cancers 2015, 7, 1758–1784.
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134.
- Iwakura, Y.; Nawa, H. ErbB1-4-dependent EGF/neuregulin signals and their cross talk in the central nervous system: Pathological implications in schizophrenia and Parkinson’s disease. Front. Cell. Neurosci. 2013, 7, 4.
- Bollu, L.; Mazumdar, A.; Savage, M.I.; Brown, P.H. Molecular Pathways: Targeting Protein Tyrosine Phosphatases in Cancer. Clin. Cancer Res. 2017, 23, 2136–2142.
- Motiwala, T.; Jacob, S.T. Role of Protein Tyrosine Phosphatases in Cancer. Prog. Nucleic Acid Res. Mol. Biol. 2006, 81, 297–329.
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein Tyrosine Phosphatases in the Human Genome. Cell 2004, 117, 699–711.
- Qu, C.K. The SHP-2 tyrosine phosphatase: Signaling mechanisms and biological functions. Cell Res. 2000, 10, 279–288.
- Eck, M.J.; Pluskey, S.; Trüb, T.; Harrison, S.C.; Shoelson, S.E. Spatial constraints on the recognition of phosphoproteins by the tandem SH2 domains of the phosphatase SH-PTP2. Nat. Cell Biol. 1996, 379, 277–280.
- Chan, R.J.; Feng, G.-S. PTPN11 is the first identified proto-oncogene that encodes a tyrosine phosphatase. Blood 2006, 109, 862–867.
- Hof, P.; Pluskey, S.; Dhe-Paganon, S.; Eck, M.J.; Shoelson, S.E. Crystal Structure of the Tyrosine Phosphatase SHP-2. Cell 1998, 92, 441–450.
- Matozaki, T.; Murata, Y.; Saito, Y.; Okazawa, H.; Ohnishi, H. Protein tyrosine phosphatase SHP-2: A proto-oncogene product that promotes Ras activation. Cancer Sci. 2009, 100, 1786–1793.
- Zhang, S.Q.; Yang, W.; Kontaridis, M.I.; Bivona, T.G.; Wen, G.; Araki, T.; Luo, J.; Thompson, J.A.; Schraven, B.L.; Philips, M.R.; et al. Shp2 Regulates Src Family Kinase Activity and Ras/Erk Activation by Controlling Csk Recruitment. Mol. Cell 2004, 13, 341–355.
- Hanafusa, H.; Torii, S.; Yasunaga, T.; Nishida, E. Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat. Cell Biol. 2002, 4, 850–858.
- Agazie, Y.M.; Hayman, M.J. Molecular Mechanism for a Role of SHP2 in Epidermal Growth Factor Receptor Signaling. Mol. Cell. Biol. 2003, 23, 7875–7886.
- Zhang, M.; Jang, H.; Nussinov, R. The structural basis for Ras activation of PI3Kα lipid kinase. Phys. Chem. Chem. Phys. 2019, 21, 12021–12028.
- Lu, Y.; Yu, Q.; Liu, J.H.; Zhang, J.; Wang, H.; Koul, D.; McMurray, J.S.; Fang, X.; Yung, W.A.; Siminovitch, K.A.; et al. Src Family Protein-tyrosine Kinases Alter the Function of PTEN to Regulate Phosphatidylinositol 3-Kinase/AKT Cascades. J. Biol. Chem. 2003, 278, 40057–40066.
- Irby, R.B.; Yeatman, T.J. Role of Src expression and activation in human cancer. Oncogene 2000, 19, 5636–5642.
- Jiao, Q.; Bi, L.; Ren, Y.; Song, S.; Wang, Q.; Wang, Y.-S. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol. Cancer 2018, 17, 36.
- Fiordalisi, J.J.; Dewar, B.J.; Graves, L.M.; Madigan, J.P.; Cox, A.D. Src-Mediated Phosphorylation of the Tyrosine Phosphatase PRL-3 Is Required for PRL-3 Promotion of Rho Activation, Motility and Invasion. PLoS ONE 2013, 8, e64309.
- Liang, F.; Liang, J.; Wang, W.-Q.; Sun, J.-P.; Udho, E.; Zhang, Z.-Y. PRL3 Promotes Cell Invasion and Proliferation by Down-regulation of Csk Leading to Src Activation. J. Biol. Chem. 2007, 282, 5413–5419.
- Wang, Y.; Yan, F.; Ye, Q.; Wu, X.; Jiang, F. PTP1B inhibitor promotes endothelial cell motility by activating the DOCK180/Rac1 pathway. Sci. Rep. 2016, 6, 24111.
- Liu, F.; Sells, M.A.; Chernoff, J. Protein tyrosine phosphatase 1B negatively regulates integrin signaling. Curr. Biol. 1998, 8, 173–S2.
- Zhao, X.; Guan, J.-L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Deliv. Rev. 2011, 63, 610–615.
- Defilippi, P.; Di Stefano, P.; Cabodi, S. p130Cas: A versatile scaffold in signaling networks. Trends Cell Biol. 2006, 16, 257–263.
- Liu, F.; Hill, D.E.; Chernoff, J. Direct Binding of the Proline-rich Region of Protein Tyrosine Phosphatase 1B to the Src Homology 3 Domain of p130Cas. J. Biol. Chem. 1996, 271, 31290–31295.
- Burridge, K.; Wennerberg, K. Rho and Rac Take Center Stage. Cell 2004, 116, 167–179.
- Brábek, J.; Constancio, S.S.; Shin, N.-Y.; Pozzi, A.; Weaver, A.M.; Hanks, S.K. CAS promotes invasiveness of Src-transformed cells. Oncogene 2004, 23, 7406–7415.
- Zhang, J.; Wu, H.; Yi, B.; Zhou, J.; Wei, L.; Chen, Y.; Zhang, L. RING finger protein 38 induces gastric cancer cell growth by decreasing the stability of the protein tyrosine phosphatase SHP-1. FEBS Lett. 2018, 592, 3092–3100.
- Joo, M.K.; Park, J.-J.; Yoo, H.S.; Lee, B.J.; Chun, H.J.; Lee, S.W.; Bak, Y.-T. Epigenetic regulation and anti-tumorigenic effects of SH2-containing protein tyrosine phosphatase 1 (SHP1) in human gastric cancer cells. Tumor Biol. 2015, 37, 4603–4612.
- Liao, S.-C.; Li, J.-X.; Yu, L.; Sun, S.-R. Protein tyrosine phosphatase 1B expression contributes to the development of breast cancer. J. Zhejiang Univ. Sci. B 2017, 18, 334–342.
- Bentires-Alj, M.; Neel, B.G. Protein-Tyrosine Phosphatase 1B Is Required for HER2/Neu–Induced Breast Cancer. Cancer Res. 2007, 67, 2420–2424.
- Karlsson, E.; Veenstra, C.; Gårsjö, J.; Nordenskjöld, B.; Fornander, T.; Stål, O. PTPN2 deficiency along with activation of nuclear Akt predict endocrine resistance in breast cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 599–607.
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643.
- Miller, T.W. Endocrine Resistance: What Do We Know? Am. Soc. Clin. Oncol. Educ. Book 2013, e37–e42.
- Klingler-Hoffmann, M.; Fodero-Tavoletti, M.T.; Mishima, K.; Narita, Y.; Cavenee, W.K.; Furnari, F.; Huang, H.-J.S.; Tiganis, T. The Protein Tyrosine Phosphatase TCPTP Suppresses the Tumorigenicity of Glioblastoma Cells Expressing a Mutant Epidermal Growth Factor Receptor. J. Biol. Chem. 2001, 276, 46313–46318.
- Tiganis, T.; Kemp, B.E.; Tonks, N.K. The Protein-tyrosine Phosphatase TCPTP Regulates Epidermal Growth Factor Receptor-mediated and Phosphatidylinositol 3-Kinase-dependent Signaling. J. Biol. Chem. 1999, 274, 27768–27775.
- Shields, B.J.; Wiede, F.; Gurzov, E.N.; Wee, K.; Hauser, C.; Zhu, H.-J.; Molloy, T.J.; O’Toole, S.A.; Daly, R.J.; Sutherland, R.L.; et al. TCPTP Regulates SFK and STAT3 Signaling and Is Lost in Triple-Negative Breast Cancers. Mol. Cell. Biol. 2013, 33, 557–570.
- Tiganis, T.; Bennett, A.M. Protein tyrosine phosphatase function: The substrate perspective. Biochem. J. 2007, 402, 1–15.
- Shields, B.J.; Hauser, C.; Bukczynska, P.E.; Court, N.W.; Tiganis, T. DNA Replication Stalling Attenuates Tyrosine Kinase Signaling to Suppress S Phase Progression. Cancer Cell 2008, 14, 166–179.
- Simoncic, P.D.; Lee-Loy, A.; Barber, D.L.; Tremblay, M.L.; McGlade, C. The T Cell Protein Tyrosine Phosphatase Is a Negative Regulator of Janus Family Kinases 1 and 3. Curr. Biol. 2002, 12, 446–453.
- Tiganis, T.; Bennett, A.M.; Ravichandran, K.S.; Tonks, N.K. Epidermal Growth Factor Receptor and the Adaptor Protein p52 Shc Are Specific Substrates of T-Cell Protein Tyrosine Phosphatase. Mol. Cell. Biol. 1998, 18, 1622–1634.
- Van Vliet, C.; Bukczynska, P.E.; Puryer, M.A.; Sadek, C.M.; Shields, B.J.; Tremblay, M.L.; Tiganis, T. Selective regulation of tumor necrosis factor–induced Erk signaling by Src family kinases and the T cell protein tyrosine phosphatase. Nat. Immunol. 2005, 6, 253–260.
- Yamada, O.; Ozaki, K.; Akiyama, M.; Kawauchi, K. JAK–STAT and JAK–PI3K–mTORC1 Pathways Regulate Telomerase Transcriptionally and Posttranslationally in ATL Cells. Mol. Cancer Ther. 2012, 11, 1112–1121.
- Wu, L.; Gao, L.; Kong, D.; Xue, H. Loss of Tyrosine Phosphatase Delta Promotes Gastric Cancer Progression via Signal Transducer and Activator of Transcription 3 Pathways. Dig. Dis. Sci. 2019, 64, 3164–3172.
- Arias-Romero, L.E.; Saha, S.; Villamar-Cruz, O.; Yip, S.-C.; Ethier, S.P.; Zhang, Z.-Y.; Chernoff, J. Activation of Src by Protein Tyrosine Phosphatase 1B Is Required for ErbB2 Transformation of Human Breast Epithelial Cells. Cancer Res. 2009, 69, 4582–4588.
- Liu, X.; Chen, Q.; Hu, X.-G.; Zhang, X.-C.; Fu, T.-W.; Liu, Q.; Liang, Y.; Zhao, X.-L.; Zhang, X.; Ping, Y.-F.; et al. PTP1B promotes aggressiveness of breast cancer cells by regulating PTEN but not EMT. Tumor Biol. 2016, 37, 13479–13487.
- Wu, C.; Zhang, L.; Bourne, P.A.; Reeder, J.E.; di Sant’Agnese, P.A.; Yao, J.L.; Na, Y.; Huang, J. Protein tyrosine phosphatase PTP1B is involved in neuroendocrine differentiation of prostate cancer. Prostate 2006, 66, 1125–1135.
- Hu, C.-D.; Choo, R.; Huang, J. Neuroendocrine Differentiation in Prostate Cancer: A Mechanism of Radioresistance and Treatment Failure. Front. Oncol. 2015, 5, 90.
- Ahlgren, G.; Pedersen, K.; Lundberg, S.; Aus, G.; Hugosson, J.; Abrahamsson, P.-A. Regressive changes and neuroendocrine differentiation in prostate cancer after neoadjuvant hormonal treatment. Prostate 2000, 42, 274–279.
- Jiborn, T.; Bjartell, A.; Abrahamsson, P.-A. Neuroendocrine Differentiation in Prostatic Carcinoma During Hormonal Treatment. Urology 1998, 51, 585–589.
- Di Sant’Agnese, P.A.; de Mesy Jensen, K.L.; Churukian, C.J.; Agarwal, M.M. Human prostatic endocrine-paracrine (APUD) cells. Distributional analysis with a comparison of serotonin and neuron-specific enolase immunoreactivity and silver stains. Arch. Pathol. Lab. Med. 1985, 109, 607–612.
- Abrahamsson, P.-A.; Wadström, L.B.; Alumets, J.; Falkmer, S.; Grimelius, L. Peptide-Hormone- and Serotonin-Immunoreactive Tumour Cells in Carcinoma of the Prostate. Pathol.-Res. Pr. 1987, 182, 298–307.
- Lee, L.-F.; Guan, J.; Qiu, Y.; Kung, H.-J. Neuropeptide-Induced Androgen Independence in Prostate Cancer Cells: Roles of Nonreceptor Tyrosine Kinases Etk/Bmx, Src, and Focal Adhesion Kinase. Mol. Cell. Biol. 2001, 21, 8385–8397.
- Lessard, L.; Labbé, D.; Deblois, G.; Bégin, L.R.; Hardy, S.; Mes-Masson, A.-M.; Saad, F.; Trotman, L.C.; Giguère, V.; Tremblay, M.L. PTP1B Is an Androgen Receptor–Regulated Phosphatase That Promotes the Progression of Prostate Cancer. Cancer Res. 2012, 72, 1529–1537.
- Zhang, Y.; Li, Z.; Fan, X.; Xiong, J.; Zhang, G.; Luo, X.; Li, K.; Jie, Z.; Cao, Y.; Huang, Z.; et al. PRL-3 promotes gastric cancer peritoneal metastasis via the PI3K/AKT signaling pathway in�vitro and in�vivo. Oncol. Lett. 2018, 15, 9069–9074.
- Wang, H.; Quah, S.Y.; Dong, J.M.; Manser, E.; Tang, J.P.; Zeng, Q. PRL-3 Down-regulates PTEN Expression and Signals through PI3K to Promote Epithelial-Mesenchymal Transition. Cancer Res. 2007, 67, 2922–2926.
- Xiong, J.; Li, Z.; Zhang, Y.; Li, D.; Zhang, G.; Luo, X.; Jie, Z.; Liu, Y.; Cao, Y.; Le, Z.; et al. PRL-3 promotes the peritoneal metastasis of gastric cancer through the PI3K/Akt signaling pathway by regulating PTEN. Oncol. Rep. 2016, 36, 1819–1828.
- Hollander, P.D.; Rawls, K.; Tsimelzon, A.; Shepherd, J.; Mazumdar, A.; Hill, J.; Fuqua, S.A.W.; Chang, J.C.; Osborne, C.K.; Hilsenbeck, S.G.; et al. Phosphatase PTP4A3 Promotes Triple-Negative Breast Cancer Growth and Predicts Poor Patient Survival. Cancer Res. 2016, 76, 1942–1953.
- Diamantopoulou, Z.; Kitsou, P.; Menashi, S.; Courty, J.; Katsoris, P. Loss of Receptor Protein Tyrosine Phosphatase β/ζ (RPTPβ/ζ) Promotes Prostate Cancer Metastasis. J. Biol. Chem. 2012, 287, 40339–40349.
- Maeda, N.; Nishiwaki, T.; Shintani, T.; Hamanaka, H.; Noda, M. 6B4 Proteoglycan/Phosphacan, an Extracellular Variant of Receptor-like Protein-tyrosine Phosphatase ζ/RPTPβ, Binds Pleiotrophin/Heparin-binding Growth-associated Molecule (HB-GAM). J. Biol. Chem. 1996, 271, 21446–21452.
- Meng, K.; Rodriguez-Pena, A.; Dimitrov, T.; Chen, W.; Yamin, M.; Noda, M.; Deuel, T.F. Pleiotrophin signals increased tyrosine phosphorylation of beta-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase beta/zeta. Proc. Natl. Acad. Sci. USA 2000, 97, 2603–2608.
- Raulo, E.; Chernousov, M.; Carey, D.; Nolo, R.; Rauvala, H. Isolation of a neuronal cell surface receptor of heparin binding growth-associated molecule (HB-GAM). Identification as N-syndecan (syndecan-3). J. Biol. Chem. 1994, 269, 12999–13004.
- Perez-Pinera, P.; Alcántara, S.; Dimitrov, T.; Vega, J.A.; Deuel, T.F. Pleiotrophin disrupts calcium-dependent homophilic cell-cell adhesion and initiates an epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2006, 103, 17795–17800.
- Keane, M.M.; Lowrey, G.A.; Ettenberg, S.A.; Dayton, M.A.; Lipkowitz, S. The protein tyrosine phosphatase DEP-1 is induced during differentiation and inhibits growth of breast cancer cells. Cancer Res. 1996, 56, 4236–4243.
- Tonks, N.K. Protein tyrosine phosphatases—From housekeeping enzymes to master regulators of signal transduction. FEBS J. 2012, 280, 346–378.
- Spring, K.; Fournier, P.; Lapointe, L.; Chabot, C.; Roussy, J.; Pommey, S.; Stagg, J.; Royal, I. The protein tyrosine phosphatase DEP-1/PTPRJ promotes breast cancer cell invasion and metastasis. Oncogene 2015, 34, 5536–5547.
- Zhang, S.; Zhang, R.; Xu, R.; Shang, J.; He, H.; Yang, Q. MicroRNA-574-5p in gastric cancer cells promotes angiogenesis by targeting protein tyrosine phosphatase non-receptor type 3 (PTPN3). Gene 2020, 733, 144383.
- Li, M.-Y.; Peng, W.-H.; Wu, C.-H.; Chang, Y.-M.; Lin, Y.-L.; Chang, G.-D.; Wu, H.-C.; Chen, G.-C. PTPN3 suppresses lung cancer cell invasiveness by counteracting Src-mediated DAAM1 activation and actin polymerization. Oncogene 2019, 38, 7002–7016.
- Aspenström, P.; Richnau, N.; Johansson, A.-S. The diaphanous-related formin DAAM1 collaborates with the Rho GTPases RhoA and Cdc42, CIP4 and Src in regulating cell morphogenesis and actin dynamics. Exp. Cell Res. 2006, 312, 2180–2194.
This entry is offline, you can click here to edit this entry!