The pathophysiology of viral myocarditis and its sequelae leading to severe heart failure with a poor prognosis is not fully understood and represents a significant public health issue globally. Most likely, at a certain point, besides viral persistence, several etiological types merge into a common pathogenic autoimmune process leading to chronic inflammation and tissue remodeling, ultimately resulting in the clinical phenotype of dilated cardiomyopathy.
The term “myocarditis” was introduced by Jean-Nicolas Corvisart in the early 19th century. The exact incidence of myocarditis (acute and chronic) remains unclear, mainly due to the heterogeneity in symptomatology ranging from nonspecific symptoms of fatigue to fulminant acute heart failure with the need for heart transplantation
[1].
Most cases of myocarditis are probably caused by infectious agents, although often, the pathogen cannot be detected after the onset of the disease. Different virus genomes have been found to be associated with myocarditis: especially a high prevalence of parvovirus B19 (B19V), member of the
Parvoviridae family genus
Erythroparvovirus and human herpesvirus 6 (HHV6), indicating a shift in the last years
[2][3][4][5] Viral persistence in the myocardium is associated with progressive deterioration of left ventricular ejection fraction (LVEF), whereas elimination of the viral genomes led to a marked improvement in left ventricular (LV) function
[43][44]. The presence of active virus replication, which has received less attention so far, probably plays a decisive role in this process
[45][46][47].
Pathogens, including viruses, can secondarily trigger autoimmune mechanisms. Most likely, at a certain point, several etiological types merge into a common pathogenic autoimmune process, leading to chronic inflammation and tissue remodeling, ultimately resulting in the clinical phenotype of dilated cardiomyopathy (DCM)
[48][49][50][51][52].
Acute myocarditis is diagnosed by histological, immunological and immunohistochemical criteria and clinically implies a short time elapsed from the onset of symptoms and diagnosis, while dilated inflammatory cardiomyopathy (DCMi) indicates myocarditis in association with cardiac dysfunction. Chronic myocarditis could represent an intermediate stage with a longer duration of symptoms (>1 month) in patients with persisting myocardial inflammation.
Endomyocardial biopsy (EMB) with histology, immunostaining for inflammation and polymerase chain reaction (PCR) remains the gold standard for the diagnosis due to its definitive capacity and etiologic diagnosis (viral or immune-mediated) in myocarditis and DCMi
[1][53][54][55]. Here, noninvasive diagnostics fail when infectious agents are involved because they cannot detect or quantify different viral types or subtypes or the degree and quality of inflammation to identify specific forms of immune response. Moreover, the use of advanced diagnostic tools has led to better identification of the etiology of myocarditis and to a new interest in the mechanisms of the inflammatory process in the heart. Understanding the underlying molecular mechanisms is necessary to assess the prognosis of patients and is fundamental to appropriate specific and personalized therapeutic strategies.
The severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) infection and the resulting clinical manifestation of coronavirus disease 2019 (COVID-19) spread rapidly throughout the world. Cardiac involvement in the current COVID-19 epidemic is reported; however, whether myocardial injury may be related to the effects of the generalized infection or direct cardiac involvement is still under investigation. Nevertheless, this is the worst pandemic in a century and has awakened a special sensitivity to viral infections throughout the world.
2. General Pathophysiological Aspects
It stands to reason that the ability to predict, with reasonable accuracy, the clinical course and progression of disease in patients with viral myocarditis would facilitate resource management and the early application of certain therapeutic options, including pharmacological treatment
[5][49][56][57]. To better understand the course of the disease and to make a prognostic statement, it is necessary to address the underlying pathophysiological process. After the acute phase of viral-mediated myocarditis, there are three commonly accepted clinical possibilities: (1) the virus is cleared without residual inflammation, resulting in complete healing; (2) the viral infection persists with or without inflammation; or (3) the viral infection results in autoimmune-mediated inflammation that persists despite clearance of the virus
[4][52][58][59].
If the infectious agent is rapidly eliminated and the inflammatory process is finalized, the disease will heal with only minor changes in the myocardium. At this stage, the true causes of the disease can no longer be determined. These patients usually recover completely within weeks to months.
In contrast, when the viral infection has been overcome, and the antiviral immune response has subsided, but irreversible myocardial damage has already developed, the clinical picture evolves into DCM. In this situation, diagnostics have started too late, and they cannot elucidate the original causes of the disease. Therefore, current data argue for the need to identify patients at an early and still reversible stage of virus-associated heart disease
[60] (
Figure 1). The aim of any diagnostics is to enable a precise diagnosis that can differentiate between virus positivity or virus exclusion, proof of inflammation, characterization and intensity. This is possible by the use of the EMB to enable a specific, personalized and causal treatment option.
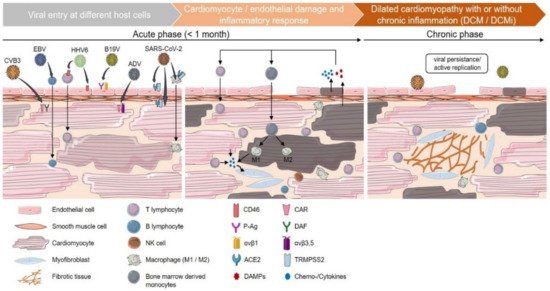
Figure 1. Phases of viral-mediated myocarditis. Cardiotropic viruses enter the myocardium via different host-cellular routes. Coxsackievirus B3 (CVB3) and adenovirus (ADV) directly target cardiomyocytes via the transmembrane coxsackievirus and adenovirus receptor (CAR). The decay-accelerating factor (DAF) serves as an additional CVB3 receptor, whereas integrins (αvβ3 and αvβ5) act as co-receptors for ADV internalization. Human herpesvirus 6 (HHV6) primarily targets CD4+ t lymphocytes and endothelial cells using CD46 as a cellular receptor. Epstein–Barr virus (EBV) enters cardiac tissue by infection of resting human b lymphocytes and subsequent infiltration into adjacent tissue. Parvovirus B19 (B19V) infects endothelial cells using erythrocyte P antigen (P-Ag) and integrin αvβ1 as a co-receptor. For SARS-CoV-2, several cardiac targets, such as cardiomyocytes, endothelial cells and circulating macrophages, are suggested. Its cellular entry depends on the expression of angiotensin-converting enzyme 2 (ACE2) and transmembrane protease serine subtype 2 (TMPRSS2) on the host-cell surface. Internalization of viral particles triggers a broad spectrum of host-cell responses and the activation of the innate immune system. Direct cardiomyocytolysis or apoptosis is induced by active viral replication and its transcription products (CVB3 and ADV). Indirectly, cardiomyocyte impairment can arise as a consequence of vascular endothelial dysfunction (B19V, HHV6). In addition, myocarditis is suggested to be induced by infected immune cells carrying viral genomes into the myocardium (HHV6, EBV). As a result of cardiac damage, inflammatory cytokines, as well as damage-associated molecular patterns (DAMPs), are released, triggering the infiltration of mononuclear cells, such as lymphocytes and monocytes, which differentiate into M1 or M2 macrophages depending on the inflammatory milieu. The further release of proinflammatory chemo and cytokines leads to the activation of heart-resident myofibroblasts and an increased generation of fibrous tissue. In the case of viral persistence, viral myocarditis can contribute to the chronic deterioration of cardiac function and the clinical presentation of dilated cardiomyopathy (DCM).
2.2. Autoimmunity in Viral Myocarditis
The chronic immune stimulation or autoimmunity in chronic viral myocarditis is the result of incompletely overcoming viral infection or a response to the preceding virus or immune-mediated chronic tissue injury. By understanding this pathophysiology, it is obvious that viral diagnostics in EMB is always required when inflammation is confirmed. Both the persistent antigenic trigger by continuously synthesized viral proteins and the release of intracellular proteins from necrotic or apoptotic myocardial cells can stimulate chronic inflammation that may eventually involve the entire myocardium.
Autoimmune reactions—possibly favored by molecular mimicry—activate virus-specific T cells that attack the myocardium. High concentrations of cytokines (e.g., tumor necrosis factor (TNF), interleukin (IL)-1a, IL-1b, IL-2 and interferon- (IFN-) γ) are produced during this phase. These cytokines, together with antibodies against viral and cardiac proteins, further exacerbate the damage to the heart and impairment of systolic function due to disturbance of the contractile apparatus and matrix proteins
[61][62][63][64][65].
Recent studies have investigated the T helper (Th) 17 effector functions during viral infections, including its critical role in the production and induction of proinflammatory cytokines and in the recruitment and activation of other immune cells. Thus, Th17 is involved in the induction of both pathogenicity and immunoprotective mechanisms observed in the host immune response to viruses and can also modulate immune responses
[48].
Genetics may play a role in predisposing certain populations to be more susceptible to infection and possibly, to develop myocarditis as an autoimmune response once infected with a cardiotropic virus. The geographic and temporal distribution of viral strains identified in the past suggests that viral strains specifically affect local populations and evolve over distance, with a seasonal distribution likely
[66].
3. Treatment Options
The definitive differentiation between virus positivity/transcriptional activity or virus exclusion, and the evidence of inflammation, its characterization and intensity are a fundamental prerequisite for a specific therapeutic decision based on EMB analyses. The first randomized trial on viral cardiomyopathy was the placebo-controlled phase II multicenter BICC-Trial (Betaferon In Chronic Viral Cardiomyopathy) [7]. Compared to placebo, in enterovirus and adenovirus infections, interferon-β-1b was leading to effective virus clearance in follow-up EMB after treatment, associated with favorable effects on NYHA functional class, improvement in quality of life and global patient assessment.
A recent study demonstrated the benefit of nucleoside analog treatment in controlling B19V replication and reducing viral transcripts, as well as rapidly improving symptoms in patients with active B19V infection [19].
Myocardial inflammation that persists after viral elimination requires immunosuppressive treatment to prevent subsequent autoimmune-mediated myocardial damage. However, viral genomes must be excluded prior to immunosuppressive therapy. Treatment for these patients with post-viral autoimmunological myocarditis consists of corticosteroids, azathioprine or ciclosporin A, in addition to optimal heart failure medication [73][89][168].
This entry is adapted from the peer-reviewed paper 10.3390/jcm10225240