The cystic fibrosis lung is inflamed and therefore the impact of inflammation on CFTR rescue should be considered. There is now evidence that airway inflammation enhances CFTR rescue. The development of CFTR modulators, such as correctors that augment F508del CFTR transfer to the apical membrane, and potentiators that increase CFTR channel activity, permitted successful treatment of the basic defect in cystic fibrosis. The first FDA-approved CFTR modulator was the potentiator ivacaftor (VX-770), which improves the function of the gating mutant G551D CFTR. While the potentiator VX-770 or the CFTR corrector lumacaftor (VX-809) alone did not significantly improve lung function in F508del CF patients, combining VX-809 with VX-770 (in the drug Orkambi) or combining the newer corrector tezacaftor (VX-661) with VX-770 (in the drug Symdeko) resulted in modest lung function improvements in clinical trials in patients homozygous for F508del CFTR. Notably, F508del rescue resulting from these combination therapies or the clinically effective novel triple therapy (in the drug Trikafta) were enhanced by airway epithelial inflammation in vitro. Thus, the airway inflammatory milieu improves the efficacy of CFTR modulators.
- CFTR
- airway inflammation
1. Evaluation of CFTR Rescue in Inflamed Airway Epithelia In Vitro
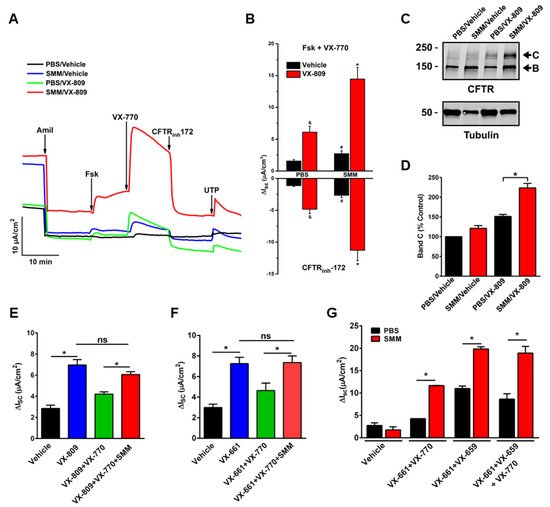
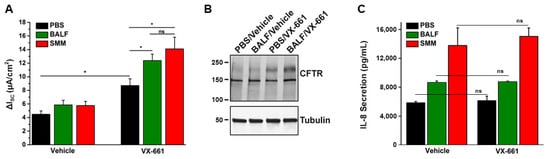
2. Airway Inflammation Enhances CFTR Modulator Therapy-Improved Lung Function In Vivo
This entry is adapted from the peer-reviewed paper 10.3390/cells10113260
References
- Bitam, S.; Pranke, I.; Hollenhorst, M.; Servel, N.; Moquereau, C.; Tondelier, D.; Hatton, A.; Urbach, V.; Sermet-Gaudelus, I.; Hinzpeter, A.; et al. An unexpected effect of TNF-alpha on F508del-CFTR maturation and function. F1000Res 2015, 4, 218.
- Brouillard, F.; Bouthier, M.; Leclerc, T.; Clement, A.; Baudouin-Legros, M.; Edelman, A. NF-kappa B mediates up-regulation of CFTR gene expression in Calu-3 cells by interleukin-1beta. J. Biol. Chem. 2001, 276, 9486–9491.
- Galietta, L.J.; Pagesy, P.; Folli, C.; Caci, E.; Romio, L.; Costes, B.; Nicolis, E.; Cabrini, G.; Goossens, M.; Ravazzolo, R.; et al. IL-4 is a potent modulator of ion transport in the human bronchial epithelium in vitro. J. Immunol. 2002, 168, 839–845.
- Danahay, H.; Atherton, H.; Jones, G.; Bridges, R.J.; Poll, C.T. Interleukin-13 induces a hypersecretory ion transport phenotype in human bronchial epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 282, L226–L236.
- Simões, F.B.; Kmit, A.; Amaral, M.D. Cross-talk of inflammatory mediators and airway epithelium reveals CFTR as a major target. ERJ Open Res. 2021.
- Cruz, D.F.; Mitash, N.; Farinha, C.M.; Swiatecka-Urban, A. TGF-beta1 Augments the Apical Membrane Abundance of Lemur Tyrosine Kinase 2 to Inhibit CFTR-Mediated Chloride Transport in Human Bronchial Epithelia. Front. Cell Dev. Biol. 2020, 8, 58.
- Pruliere-Escabasse, V.; Fanen, P.; Dazy, A.C.; Lechapt-Zalcman, E.; Rideau, D.; Edelman, A.; Escudier, E.; Coste, A. TGF-beta 1 downregulates CFTR expression and function in nasal polyps of non-CF patients. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, L77–L83.
- Snodgrass, S.M.; Cihil, K.M.; Cornuet, P.K.; Myerburg, M.M.; Swiatecka-Urban, A. Tgf-beta1 inhibits Cftr biogenesis and prevents functional rescue of DeltaF508-Cftr in primary differentiated human bronchial epithelial cells. PLoS ONE 2013, 8, e63167.
- Stanton, B.A.; Coutermarsh, B.; Barnaby, R.; Hogan, D. Pseudomonas aeruginosa Reduces VX-809 Stimulated F508del-CFTR Chloride Secretion by Airway Epithelial Cells. PLoS ONE 2015, 10, e0127742.
- Gentzsch, M.; Cholon, D.M.; Quinney, N.L.; Martino, M.E.B.; Minges, J.T.; Boyles, S.E.; Guhr Lee, T.N.; Esther, C.R., Jr.; Ribeiro, C.M.P. Airway Epithelial Inflammation In Vitro Augments the Rescue of Mutant CFTR by Current CFTR Modulator Therapies. Front. Pharmacol. 2021, 12, 628722.
- Gentzsch, M.; Cholon, D.M.; Quinney, N.L.; Boyles, S.E.; Martino, M.E.B.; Ribeiro, C.M.P. The cystic fibrosis airway milieu enhances rescue of F508del in a pre-clinical model. Eur. Respir. J. 2018, 52, 1801133.
- Cholon, D.M.; Quinney, N.L.; Fulcher, M.L.; Esther, C.R., Jr.; Das, J.; Dokholyan, N.V.; Randell, S.H.; Boucher, R.C.; Gentzsch, M. Potentiator ivacaftor abrogates pharmacological correction of DeltaF508 CFTR in cystic fibrosis. Sci. Transl. Med. 2014, 6, 246ra296.
- Rehman, T.; Karp, P.H.; Tan, P.; Goodell, B.J.; Pezzulo, A.A.; Thurman, A.L.; Thornell, I.M.; Durfey, S.L.; Duffey, M.E.; Stoltz, D.A.; et al. Inflammatory cytokines TNF-alpha and IL-17 enhance the efficacy of cystic fibrosis transmembrane conductance regulator modulators. J. Clin. Investig. 2021, 131, e150398.
- Ribeiro, C.M.; Paradiso, A.M.; Schwab, U.; Perez-Vilar, J.; Jones, L.; O’Neal, W.; Boucher, R.C. Chronic airway infection/inflammation induces a Ca2+i-dependent hyperinflammatory response in human cystic fibrosis airway epithelia. J. Biol. Chem. 2005, 280, 17798–17806.
- Ribeiro, C.M.; Paradiso, A.M.; Carew, M.A.; Shears, S.B.; Boucher, R.C. Cystic fibrosis airway epithelial Ca2+ i signaling: The mechanism for the larger agonist-mediated Ca2+ i signals in human cystic fibrosis airway epithelia. J. Biol. Chem. 2005, 280, 10202–10209.
- Martino, M.E.; Olsen, J.C.; Fulcher, N.B.; Wolfgang, M.C.; O’Neal, W.K.; Ribeiro, C.M. Airway epithelial inflammation-induced endoplasmic reticulum Ca2+ store expansion is mediated by X-box binding protein-1. J. Biol. Chem. 2009, 284, 14904–14913.
- Ribeiro, C.M.; O’Neal, W.K. Endoplasmic reticulum stress in chronic obstructive lung diseases. Curr. Mol. Med. 2012, 12, 872–882.
- Gentzsch, M.; Ren, H.Y.; Houck, S.A.; Quinney, N.L.; Cholon, D.M.; Sopha, P.; Chaudhry, I.G.; Das, J.; Dokholyan, N.V.; Randell, S.H.; et al. Restoration of R117H CFTR folding and function in human airway cells through combination treatment with VX-809 and VX-770. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L550–L559.
- Veit, G.; Avramescu, R.G.; Perdomo, D.; Phuan, P.W.; Bagdany, M.; Apaja, P.M.; Borot, F.; Szollosi, D.; Wu, Y.S.; Finkbeiner, W.E.; et al. Some gating potentiators, including VX-770, diminish DeltaF508-CFTR functional expression. Sci. Transl. Med. 2014, 6, 246ra297.