Bile acids are digestive agents synthesized in the liver and released into the gastrointestinal track during normal physiological conditions to aid in the emulsification of lipids and fat-soluble vitamins. These bile acids are highly regulated via enterohepatic circulation, a process which minimizes bile acid loss through a wide network of organs and bile acid transporters. Beyond this, bile acids and the activation of bile acid receptors has been observed in extrahepatic tissues, in particular the central nervous system. Certain bile acids, through the use of specific bile acid transporters, can also gain entry into the brain via the blood brain barrier. Furthermore, there has been an increase in recent literature highlighting bile acids and the presence of bile acid signaling in the brain and neural cells. This entry is a current overview of these topics.
- Bile acids
- Bile acid synthesis
- Bile acid transporters
- Bile acid receptors
- Enterohepatic circulation
- Central nervous system
- FXR
- TGR5
1. Introduction
Bile acids are amphipathic molecules synthesized in the liver, stored in the gallbladder and released into the intestinal lumen in response to food intake as a digestion mechanism. Their primary function is to serve as detergents in the solubilization of dietary lipids and fat-soluble vitamins. The majority of bile acids are passively or actively recovered throughout the intestinal tract and then returned to the liver for recycling via enterohepatic circulation with a small percentage of bile acids excreted as waste. Bile acids maintain secondary functions as steroid hormones and influence metabolic processes as potent signaling molecules via membrane-bound receptors such as sphingosine-1-phosphate receptor 2 (S1PR2) and Takeda G-protein-coupled bile acid receptor 5 (TGR5) and nuclear receptors such as Farnesoid X receptor (FXR) [1]. Surprisingly, emerging evidence suggests that bile acid signaling may also play a role in the physiology and pathophysiology of the brain. Furthermore, the usage of the bile acids ursodeoxycholic acid (UDCA) and tauroursodeoxycholic acid (TUDCA) may possess therapeutic benefits in neurological ailments due to their neuroprotective properties, lack of cytotoxicity and permeability across the blood brain barrier (BBB), shown with UDCA in clinical studies [2] and TUDCA in animal models [3]. For some neurodegenerative diseases clinical trials implementing bile acid treatments may offer therapeutic potential, from a phrase III trial with UDCA [4] and to follow-up tracking of long term chenodeoxycholic acid (CDCA) efficacy [5]. Furthermore, in neurological disorders, there have been an increased amount of published studies implementing the use of bile acids or specifically targeting bile acid signaling.
2. Bile Acids Synthesis, Metabolism and Enterohepatic Circulation
Each day roughly 500mg of cholesterol is converted into bile acids in the adult human liver. There are two major bile acid synthetic pathways: the classic (or neutral) pathway that occurs in the liver and the alternate (or acidic) pathway found in peripheral tissues and the liver. A pathway for cholesterol regulation in the brain, the neural cholesterol clearance pathway, was discovered more recently and will be discussed at length below. In humans, cholic acid (CA) and CDCA are the only primary bile acids synthesized [6]. For rodents, their bile acid pool composition consists of primary bile acids CA, CDCA and the creation of α-muricholic acid (MCA) and β-muricholic (β-MCA) acid from CDCA [7]. Studies have reported sex differences in bile acid metabolism in healthy humans, with men displaying a higher percentage of fasting plasma concentrations of individual bile acids and total bile acids, increased by 111% and 51%, respectively [8]. Serum comparisons for bile acid profiles even showed significantly lower amounts of primary bile acids CA and CDCA when comparing women to men [9]. The variance of these findings with circulating bile acids may be useful when therapeutic drugs are being implemented for clinical trials.
The conversion of cholesterol to primary bile acids is facilitated by a family of unique cytochrome P450 enzymes that are located in the cytosol, endoplasmic reticulum, mitochondria, and peroxisomes. Expressed solely in the hepatocytes, the classic bile acid synthesis pathway is initiated via 7α-hydroxylase (CYP7A1) converting cholesterol into 7α-hydroxycholesterol with the resulting metabolic products of primary bile acids synthesized to CA via sterol 12α-hydroxylase (CYP8B1) or CDCA by sterol 27-hydroxylase (CYP27A1) [10]. In contrast, the alternative pathway catabolizes cholesterol in all tissues; cholesterol is metabolized via mitochondrial CYP27A1, converting it into 27-hydroxycholesterol. For further conversion, these midpoint metabolites are transported from peripheral tissues back to the liver to be converted to primary bile acids CA and CDCA [11]. The classic pathway is the primary route for bile acid synthesis regulated by CYP7A1, the only rate-limited enzyme in all bile acid synthesis. More than 90% of total bile acid production in humans is sourced from this pathway, with less than 10% of the total bile acids coming from the alternative pathway during routine physiological conditions [7]. In contrast, in healthy wild type mice, only 60% of their total bile composition is sourced from the classical pathway due to their bile acid pool, including the addition of MCA and β-MCA that are not present in healthy humans [12]. Enterohepatic circulation allows the total bile salt pool to undergo 4–12 cycles a day, an efficient method of reabsorption and recycling that ensures minimal bile acid loss via urinary or fecal excretion [13]. A summary of the bile acid synthesis pathways can be seen in Figure 1.
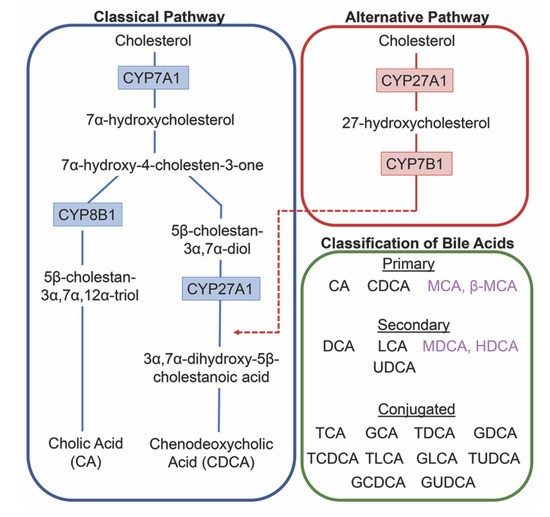
Figure 1. Bile acid synthesis pathways. The classic pathway for bile acid synthesis occurs in the hepatocytes of the liver via 7α-hydroxylase (CYP7A1) converting cholesterol into 7α-hydroxycholesterol. Primary bile acid cholic acid (CA) is formed after subsequent conversions from sterol 12α-hydroxylase (CYP8B1) and chenodeoxycholic acid from sterol 27-hydroxylase (CYP27A1). In the alternative or acidic pathway, mitochondrial CYP27A1 in peripheral tissues convert cholesterol into 27-hydroxycholesterol. Oxysterol 7α-hydroxylase (CYP7B1) is an additional assisting enzyme in this pathway and the resulting products feed back into the liver, indicated by the red arrow feeding into the classical pathway under CYP27A1. Primary and secondary bile acids specific to rodents are listed in purple. Bile acids can become conjugated with glycine or taurine after interactions with gut flora.
Before de novo primary bile acids CA and CDCA are released from the liver, some are conjugated with either glycine (in humans) or taurine (in mice), granting increased water solubility and reduced cytotoxicity to fulfill their dietary roles of lipid emulsification throughout the intestines. Taurocholic acid (TCA) and glycocholic acid (GCA) are synthesized from CA, taurochenodeoxycholic acid (TCDCA) and glycochenodeoxycholic acid (GCDCA) are synthesized from CDCA. Along with CA and CDCA, these newly synthesized conjugated bile acids are transported from hepatocytes to the bile canaliculus via the bile salt export pump (BSEP) and multidrug resistance-associated protein 2 (MRP2) for storage in the gallbladder awaiting their release into the intestinal lumen with the intake of food. Once nutrients enter the stomach, they trigger the gallbladder to release bile acids into the duodenum where they contribute to the digestion of lipids and fat-soluble vitamins. As bile acids continue through to the ileum, unconjugated and some glycine-conjugated bile acids will be reabsorbed via passive diffusion in the jejunum and colon with the majority of conjugated bile acids requiring active reabsorption via the apical sodium dependent bile acid transporter (ASBT) in the ileum. Other active membrane transporters sodium taurocholate cotransporting polypeptide (NTCP) and organic anion transport polypeptide (OATP) in hepatocytes mediate in bile acid reuptake once they’ve entered portal venous circulation [14].
Unconjugated primary bile acids not passively reabsorbed will interact with the intestinal bacteria flora present in the colon, creating secondary bile acids deoxycholic acid (DCA) from CA and lithocholic acid (LCA) and UDCA from CDCA for humans [15], with the secondary bile acids of murideoxycholic acid (MDCA) and hyodeoxycholic acid (HDCA) for mice [16]. When these secondary bile acids are recirculated back to the liver, conjugation with glycine or taurine can further differentiate them, such as the addition of taurine to UDCA forms TUDCA [17]. The enterohepatic circulation efficiently reclaims approximately 95% of bile acids and minimizes fecal and urinary bile acid expulsion with the help of a collective transporter process. Located on the membranes of ileocytes, proximal renal tubule cells and cholangiocytes, ASBT facilitates the absorption of the majority of bile acids lacking passive diffusion qualifications or reclaims bile acids in systemic circulation for portal venous distribution back to the liver, minimizing excretion in urine. The heteromeric organic solute transporter (OST) α and β located in the cytosol of renal proximal tubule cells, ileocytes and hepatocytes direct bile acids to systemic circulation [18], a process which when malfunctioning could exacerbate elevated systemic bile acids levels affecting the blood brain barrier [19,20]. A graphic depiction of the enterohepatic circulation of bile acids is shown in Figure 2.
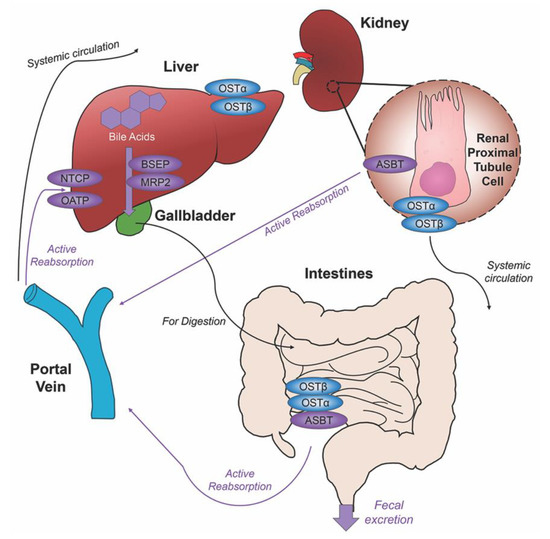
Figure 2. Enterohepatic circulation of bile acids. After primary bile acids are synthesized in the liver, the bile acid transporters bile salt export pump (BSEP) and multidrug resistance-associated protein 2 (MRP2) facilitate their storage in the gallbladder, indicated via thick purple arrow, to be released in the intestines to aid in the digestion of food. Following food intake, bile acids are released into the duodenum for the digestion of lipids and fat-soluble vitamins, bile acid movement indicated via black arrow. Some bile acids can be reabsorbed through passive diffusion in the jejunum and colon throughout the journey, while the majority of conjugated bile acids can interact with the apical sodium dependent bile acid transporter (ASBT) in the ileum for active reabsorption, indicated by multiple purple arrows. Other bile acid transporters sodium taurocholate cotransporting polypeptide (NTCP) and organic anion transport polypeptide (OATP) expressed in hepatocytes mediate active reabsorption back to the liver. Bile acids in systemic circulation will be reabsorbed by ASBT in the renal proximal tubule cells of the kidney and directed back to the liver via the portal vein. Heteromeric organic solute transporter (OST) α and β in renal proximal tubule cells, ileocytes and hepatocytes direct bile acids into systemic circulation. The efficiency of this system recycles and minimizes fecal and urinary bile acid loss by excretion.
Bile acids can undergo an additional elimination pathway consisting of glucuronidation, a process that converts hydrophobic bile acids into excretable metabolites. Uridine 5′-diphosphate-glucuronosyltransferase (UDP-glucuronosyltransferase, UGT) are multigenic enzymes that catalyze the glucuronidation reaction, conjugating glucuronic acid with exogenous and endogenous molecules. Aiding in bile acid detoxification, the resultant hydrophilic glucuronide products possess increased ability for urinary excrement [21]. Glucuronidation of bile acids leads to the important introduction of a negative charge to the molecule, allowing transport by conjugate-transporters that can facilitate bile acid-glucuronide secretion. Multidrug resistance-associated protein 1 (MRP1) and 3 (MRP3) expressed across the basolateral hepatocyte membrane aid in the efflux of glucuronides [22]. Bile acid glucuronides are present in hepatic dysfunction, with increased concentrations of glucuronidated bile acids CDCA and LCA in the plasma of patients with hepatobiliary diseases [23]. In biliary obstruction patients whose bile flow had been restored via stenting, the urinary composition of bile acid glucuronides was increased [24].
Several UGT genes have been identified in human, mouse, rat and other mammalian species. The gene superfamily consists of four UGT families, UGT1, UGT2, UGT3 and UGT8, with enzymes of UGT1 and UGT2 families the most efficient at glucuronic acid transfer [25]. Of the 18 UGT enzymes, three enzymes can be attributed to glucuronidation of bile acids: UGT2B4 for bile acids such as HDCA, UGT2B7 for primary, secondary and hydroxylated bile acids, and lastly UGT1A3 for bile acids such as CDCA, LCA, and HDCA [26]. While many UGT isoforms are predominately expressed in the liver, these enzymes are also expressed in a variety of extrahepatic tissues including the small intestines, colon, bladder, kidney, ovaries, uterus, testis, and stomach [27]. UGT expression levels and glucuronidation activity have been detected in all nine regions of the rat brain [28] and are present in the human brain [29]. Lastly, UGT has been identified in several neural cell types: neurons [30], astrocytes [30,31] and microglia [32].
3. Bile Acids in the Brain
Cholesterol is an essential component of neural development and in the composition of neurons and neuroglia, with nearly 25% of the total body cholesterol found in the brain [33]. It is a major component of the lipid molecules in the membranes of neuron and glial cells, a large fraction in the myelination performed by oligodendrocytes and is involved in the synthesis of steroid hormones [34]. While local cholesterol biosynthesis is observed at higher rates in glial cells than neurons [35], neurons solely possess the ability for cholesterol clearance. The last alternative pathway for bile acid synthesis is in the brain (shown in Figure 3), catalyzing cholesterol by neuron-specific sterol 24-hydroxylase (CYP46A1) and converting this into 24(S)-hydroxycholesterol; the increased solubility of this intermediate allows for efflux from neural tissue via the BBB through lipoprotein transport ATP-binding cassette transporter 1 (ABCA1) [36]. Specific bile acid transporters allowing the influx of bile acids from the periphery into the CNS also exist. For example, OATPs, with rat OATP1 expressed in the choroid plexus and rat OATP2 highly expressed at the BBB allow for the influx of bile salts and a variety of other amphipathic organic compounds into the CNS [37]. Similarly, subpopulations of neurons, particularly in the hypothalamus express the transported ASBT which facilitates the internalization of bile acids into neurons where they have been shown to influence the activity of the hypothalamic–pituitary–adrenal (HPA) axis [38,39].
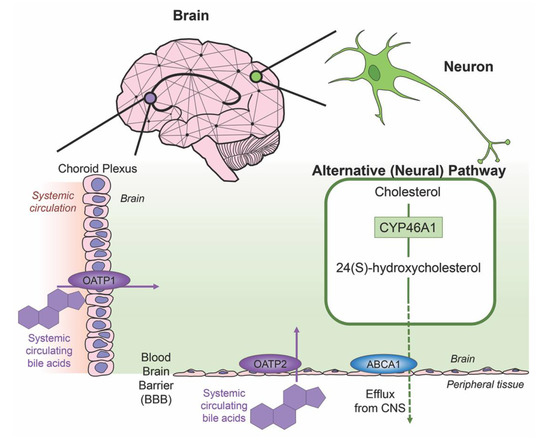
Figure 3. Neural cholesterol clearance pathway and bile acid transport into the CNS. Cholesterol is catalyzed in the brain via sterol 24-hydroxylase (CYP46A1), an enzyme expressed only in neurons. It is converted to 24(S)-hydroxycholesterol and is able to be removed from the CNS through the blood brain barrier (BBB) via the transporter ATP-binding cassette transporter 1 (ABCA1), indicated via green arrow. Other transporters mediate systemic circulating bile acids into the CNS. Organic anion transporter polypeptide 1 (OATP1) expressed in the choroid plexus and organic anion transporter polypeptide 2 (OATP2) expressed at the BBB both mediate the transport of bile acids, both processes indicated by purple arrows.
Bile acid functionality increases when acting as ligands for nuclear receptors farnesoid X receptor (FXR), the pregnane X receptor (PXR), the vitamin D receptor (VDR), and membrane receptors Takeda G-protein-coupled receptor 5 (TGR5; a G-protein-coupled receptor also called G-protein-coupled bile acid receptor 1, GPBAR-1), sphingosine-1-phosphate receptor 2 (S1PR2) and α5β1 integrin. These receptors are highly expressed in the liver and the intestines but also display activity in a variety of tissues throughout the body including the brain [40]. Bile acids can act as signaling molecules to modulate their own homeostasis. Among the bile acid receptors, FXR plays many important roles in the regulation mechanisms of bile acid synthesis and transport. FXR activation via bile acids can induce the expression of BSEP [41,42], regulating the canalicular secretion of bile acids into bile. Key players in bile acid synthesis, CYP7A1 and CYP27A1, can be repressed by FXR [22] and human UGT2B4, involved in the conversion of hydrophobic bile acids to their less toxic glucuronide derivatives, can be upregulated by FXR [43].
The affinities of primary, secondary and conjugated bile acids with individual bile acid receptors vary. Bile acids can serve as weak activators of the glucocorticoid receptor (GR) in the brain to influence the HPA axis [39]. LCA, a hydrophobic and cytotoxic bile acid, has been shown as a weak ligand for FXR with the ability to decrease BSEP expression [44]. This same bile acid is the most potent bile acid for TGR5 [45], displaying anti-tumor effects in human neuroblastoma cell cultures [46] as well as pro-apoptotic effects in breast cancer cells [47]. The primary bile acid CDCA is the most potent activator for FXR, with CA and secondary bile acids DCA and LCA showing less activation [48]. Both nuclear receptors PXR [49] and VDR [50] can be activated by secondary bile acid LCA. There are even conjugated bile acids with selective activity for receptors, such as TUDCA for α5β1 integrin [51] and TCA for S1P2R [52]. Other reviews have eloquently covered the liver and intestinal focused signaling of these receptors [11,53,54,55]. Given these points, Table 1 lists bile acid-mediated receptors that are relevant in the CNS.
Table 1. Bile Acid Receptors in the CNS.
| Receptor | Bile Acid Ligands | Cellular Localization | Expression/Functionality | References |
|---|---|---|---|---|
| FXR | CDCA, CA, DCA, LCA | Cortical neurons | Nuclear and cytoplasmic expression in cortical neurons; transcriptional activity via SHP activation. FXR deletion elevates cerebellar neurotransmitter concentrations. FXR modulates cholesterol metabolism in a rodent model of type A hepatic encephalopathy. | [56,57,58] |
| TGR5 | LCA, DCA, CDCA, CA | Neurons, astrocytes, microglia | Response to neurosteroids resulting in increased intracellular cAMP. TGR5 signaling is neuroprotective and diminishes inflammation against CCL2 in a rodent model of type A hepatic encephalopathy | [59,60] |
| S1P2R | TCA, GCA, TDCA, GDCA, TUDCA | Cortical neurons, microglia, hippocampal pyramidal cells, retinal ganglion cells | Mediates synaptic neuroplasticity, repair and neurite outgrowth. TCA activation promotes inflammation in a type A rodent model of hepatic encephalopathy | [61,62] |
| PXR | LCA | Brain endothelial cells, hippocampal neurons | BBB regulation via ABC-transporters, nonyphenol toxicity activates PXR-mediated apoptosis and neurotoxicity | [63,64] |
| VDR | LCA | Neurons, glia | Location of VDR indicates involvement with neurosteroids, confirmation of nuclear location | [65,66] |
| α5β1 integrin | TUDCA, norUDCA (UDCA homolog) | Cortical neurons, brain endothelial cells | Regulates neural morphology and migration during development, α5 influence BBB permeability | [67,68,69] |
| GR | UDCA, TCA, GCDCA, TUDCA | Neurons, microglia, cortical neurons | UDCA-bound GR modulates NF-κB-dependent transcription, GR-signaling in ginseng has protective implications in neurodegenerative models, GR-mediated HPA axis suppression is induced via injection of bile acids, GR attenuates amyloid-beta-induced apoptosis in cortical neurons through TUDCA | [39,70,71,72] |
This entry is adapted from the peer-reviewed paper 10.3390/ijms21175982