Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Physiology
The growing interest in the role of microglia in the progression of many neurodegenerative diseases is developing in an ever-expedited manner, in part thanks to emergent new tools for studying the morphological and functional features of the CNS. The discovery of specific biomarkers of the microglia phenotype could find application in a wide range of human diseases, and creates opportunities for the discovery and development of tailored therapeutic interventions.
- microglia
- potassium channel
- CNS
- neuroinflammation
- neurodegenerative disease
1. Introduction
Neurodegenerative diseases (NDDs) are disorders characterized by the progressive degeneration of the structure and function of the central or peripheral nervous systems. Recent research recognizes NDD, such as Alzheimer’s disease (AD), Huntington’s disease (HD), Parkinson’s disease (PD), frontotemporal dementia (FTD) and Amyotrophic Lateral Sclerosis (ALS) as the major causes of disability and the second leading cause of death worldwide [1]. Most NDDs are associated with a dysregulated immune response that impairs the CNS balance. Furthermore, neuroinflammation is an essential factor contributing to neurodegeneration, with reactive microglia playing a key role [2,3]. Microglia are the principal resident immune cells of the brain that support the physiological functions of the CNS. [4,5]. Furthermore, in the healthy CNS, microglia, which represent approximately 5–12% of CNS cells, are necessary for proper brain development, providing trophic support to neurons, removing apoptotic cell debris and regulating neuronal and synaptic plasticity [6,7,8,9]. Microglia are dynamic cells that shape their phenotype in a time- and context-dependent manner. In response to brain injury, microglia rapidly become reactive, assuming a detrimental or protective role.
Microglial cells were historically described as assuming two opposite activation phenotypes, including the classical M1-like pro-inflammatorystate, and the alternative, M2-like, anti-inflammatory, state [10]. Now, evidence has shown that the M1/M2 model is inadequate to describe microglia activation in vivo [11]. Indeed, microglia show multiple phenotypes associated with aging, neurodegenerative diseases and disease stage. In pathological conditions microglia can (i) rapidly change their morphology and phenotype (ii) alter the transcriptional profile and the phagocytic activity; and (iii) modify their homeostatic functions [12,13,14,15,16]. In this way, microglia can play a detrimental role in the progression of several NDDs, expressing and secreting several receptors, cytokines and chemokines that recruit additional cells and clear toxic agents and cell debris, to maintain CNS homeostasis [17,18,19,20,21]. Highlighting the complexity of microglial functions in the scenario of NDDs, it was reported that microglial cells play different roles depending on the disease and on the stage of the pathology. Microglial cells mediate host defenses against danger signals, including pathogens and proteins, such as β-Amyloid (Aβ), mutant huntingtin, prions (PrPsc), and mutant or oxidized superoxide dismutase (SOD) [22,23,24]. At first, in response to pathological brain insults, microglia initiate a defense program to restore brain homeostasis via the expression and activities of microglial pattern recognition receptors (PRRs), including toll-like receptors (TLRs), scavenger receptors (SRs), and complement receptor 3 (CR3) [25,26,27,28,29,30,31,32]. Otherwise, when the perturbations persist, microglia can assume a detrimental phenotype, initiating an exaggerated inflammatory response, resulting in neurotoxicity and neurodegeneration. Accordingly, reactive detrimental microglia were found in the brain and spinal cord of NDDs patients at advanced stages of pathology [33,34]. For example, in ALS, activated microglia have been identified in close proximity to degenerating motor neurons, suggesting a direct neurotoxic function [35]. Using transcriptome profiling of disease-associated and homeostatic genes in microglia from the brain of the 5XFAD mouse model of AD, it was possible to identify a subpopulation defined as disease-associated microglia (DAM) [36]. DAM display downregulation of homeostatic microglial genes and the upregulation of genes involved in lysosomal, phagocytic, and lipid metabolism pathways, including several known disease risk factors, such as Apolipoprotein E (Apoe), Cystatin F(Ctsd 7), lipoprotein lipase (Lpl), Tyroprotein tyrosine kinase binding protein (Tyrobp), and triggering receptor expressed on myeloid cells 2 (Trem2) [36]. These DAMs are also found in other mouse models of neurodegeneration, including the tauopathy model Tau P301S, the SOD1G93A model of ALS, the experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis (MS) and also during aging [37,38,39,40].
2. Potassium Channels: Key Regulators of Microglial Function
Among the families of ion channels, the one of potassium (K+) channels represents the most abundant group, being involved in a multitude of physiological functions in both excitable and non-excitable cells [41]. Potassium channels have transmembrane helices (TMs) spanning the lipid bilayer [42], and regulate the flow of K+ ions across the cell membrane in different cells, such as neurons, glia, and lymphocytes [41].
2.1. Potassium Channels Expressed by Microglia
Many studies in vitro and in vivo/ex vivo demonstrated that human and rodent microglia express various types of K+ channels. Pioneer research on brain acute slice or primary cultures revealed that rodent microglia express voltage-gated (Kv1.2, Kv1.3, Kv1.1, Kv1.5, Kv3.1), Ca2+-activated (KCa3.1, KCa2.3, KCa1.1), and inward rectifying K+ channels (Kir2.1) [43]. More recently, the two-pore domain channel THIK-1 (K2P13.1) has been identified as a rodent microglia signature gene and is the main K+ channel in “resting” microglia [44]. On the other hand, it has been shown that Kv1.3, KCa3.1 and Kir2.1 are the main channels expressed by unstimulated mouse neonatal cultured microglia [45].
The expression of potassium channels in human samples has been mainly demonstrated by molecular and immunohistochemistry techniques in primary cultures and post-mortem brain tissues. Studies conducted on human fetal microglia cultures have shown the presence of a higher density of K+ currents with the biophysical and pharmacological components of Kv1.3; while KCa3.1 currents were found on microglia from adult human neocortical tissue [46,47,48,49]. Kv1.3, KCa3.1 and Kir2.1 expression has been reported on post-mortem human tissue [47].
In CNS, some microglial potassium channels are also expressed by other neuronal cell types. For instance, KCa3.1 (Kcnn4) is expressed by transformed cells [50] and, in a model of spinal cord injury, it has been detected also on neurons and astrocytes [51]. Furthermore, Kv1.3 (Kcna3) is found in addition to microglia, also in astrocytes, neurons of the olfactory system, and in gliomas [52,53,54,55].
2.2. Potassium Channels Modulation in Microglia
Potassium channel expression/activation can be regulated by many stimuli in vitro and in vivo, which make them promising targets in the treatment of several pathologies that their alteration correlates with.
Among pro-inflammatory stimuli the use of the TLR-4 ligand lipopolysaccharide (LPS) predominates in microglia studies. Hankyoung and colleagues reported for the first time that LPS treatment induced expression and function of Kv1.5 and to a lesser extent Kv1.3 channels in rat primary cultures [56]. More recently, Nguyen et al. showed that pro-inflammatory or anti-inflammatory stimuli induced a different pattern of K+ channel expression in cultured neonatal mice and fetal human microglia [48]. The authors reported that neonatal mouse microglia stimulated with the LPS exhibited high KV1.3 current densities and virtually no KCa3.1 and Kir currents, while IL-4 stimulated microglia exhibited Kir2.1 currents and down-regulated KV1.3 and KCa3.1 expression [48]. On the other hand, it has been reported that IL-4 up-regulated Kcnn4 mRNA expression and increased Kca3.1 current in rat and mouse neonatal cultured microglia [57,58]. However, Lam et al. have demonstrated the existence of many different responses of rat and mouse microglia to pro- and anti-inflammatory cytokines, also in terms of K channel expression [59]. Differences have been also reported comparing functional K channel expression in adult human and rodent microglia studies [49]. In vitro studies have reported an increased expression of Kv1.1 and Kv1.2 channels in microglia upon activation with ATP [60]. Other stimuli, such as gamma interferon (γ-IFN) or granulocyte macrophage colony stimulating (GMC-S) factor induced potassium currents in microglia cells, mostly those that are voltage-gate related. [61,62,63,64].
2.3. Cellular Functions Modulated by Microglial Potassium Channels
Upon their stimulation, microglial potassium channels regulate many cellular functions such as cell volume changes, membrane potential, hormone secretion, calcium signaling, gene expression and action potential firing [41,65]. By regulating membrane potential, cell volume and intracellular ion concentration, microglial K+ channels can affect proliferation, migration toward chemotactic stimuli, phagocytosis, morphology, and respiratory bursts [66]. Specifically, in microglia, K channel activation induces membrane hyperpolarization, which drives Ca2+ influx through inward rectifying Ca2+-Release-Activated-Ca2+ channels (CRAC) [45,67], ATP-activated P2X receptors [68] and other Ca2+-permeable cation channels [67]. Therefore, K+ channels facilitate the refilling of intracellular Ca2+ stores, thus maintaining a high Ca2+ content and the timing of intracellular signaling, and playing a crucial role in cell activation and proliferation [66,69]. Although this mechanism is presented as fully proved, very little is known about microglial Ca2+ signaling in situ or in vivo, both in the healthy and in the diseased brain.
The main reason is technical in nature, as microglia largely resisted in vivo labelling with small molecules to perform calcium imaging. However, today it is possible to study the link between potassium channel activation and calcium signaling directly thanks to new in vivo tools, such as genetically-encoded Ca2+ indicators (GECI) and high resolution two-photon microscopy [70,71,72]. Focusing on the functional expression of KCa3.1 in microglia, it has been reported that these channels mainly contribute to the Ca2+ activated K+ currents in the cell [73,74] and are involved in many cell functions. KCa3.1 channels are involved in microglial migration, with cAMP/ PKA- and Ca2+ −dependent mechanisms [57,75], and in the production of reactive oxygen species (ROS) through the p38/MAPK and cGMP/PKG pathways [57,76]. Interestingly, KCa3.1 and Kv1.3 channels are involved in tumor/M2 polarized microglial cell activities, such as migration, phagocytosis, and gene expression modulation [77,78,79]. Concerning the Kv1.3 channel, it has been demonstrated that this channel played a role in proliferation of freshly isolated hippocampal microglia [80]. Crucial microglia functions in brain homeostasis such as synaptic pruning, immunosurveillance and cytokine release are regulated by tonically active THIK-1 K+ channels expressed on cell plasma membranes. In particular, Izquierdo et al., demonstrated that THIK-1, using THIK-1–blocking drugs and THIK-1–deficient mice, regulates the removal of functional excitatory synapses by promoting microglial phagocytosis of synaptic material in the developing brain [81]. Considering the involvement of K+ channels in microglial function and morphology [66,82], we aim to summarize their contribution to the progress and development of neurological disorders, as follows.
3. Potassium Channels: Implications in NDDs
K+ channels have been involved in inflammation-mediated neurotoxicity [83]. The nature of the noxious stimuli in the CNS and their chronic production affects microglial functions, potentially leading to an exaggerated proinflammatory response, neurotoxicity, and neurodegeneration. In the last few years, several studies proposed to use K+ channel inhibitors to modulate microglial activation, to reduce inflammation in NDDs (Figure 1). Expression and functional alterations of K+ channels cause neuronal dysfunction and affect membrane excitability, contributing to the progress and development of many NDDs, such as AD, PD, ALS and other pathological conditions [84,85,86,87,88,89]. Two K+ channels, the calcium-activated KCa3.1 and the voltage-gated Kv1.3, play important roles in microglia activation by modulating Ca2+ signaling and membrane potential in CNS diseases. Because these channels are widely expressed and are implicated in immune cell activation, Kv1.3 and KCa3.1 inhibitors have been studied as potential targets for disease treatment. Before going on to review the role of microglia potassium channels in murine and human models of NDDs, it is important to mention that (i) several works observed critical differences between human and mouse microglia, especially in the context of aging and neurodegenerative conditions [90,91]; (ii) since the isolation of microglia presents several barriers, as microglia are highly sensitive to manipulations, slice cultures are recently used for human tissue to better preserve the environment. Furthermore, new techniques allow microglia phenotype characterization in live brain tissue as demonstrated in the work of Milior and colleagues [92].
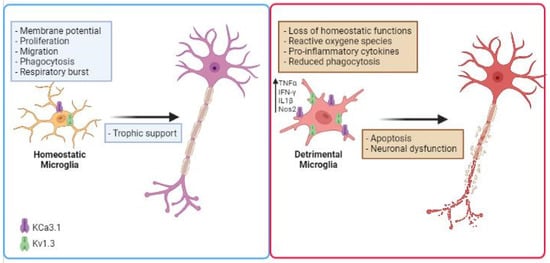
Figure 1. Schematic illustration describing the role of microglial K+ channels in NDD. Microglial cells increase the expression/activity of Kv1.3 and KCa3.1 in several neurodegenerative diseases, losing their homeostatic functions. This is associated with the release of inflammatory cytokines such as IFN-γ and TNF-α, production of noxious substances (ROS), reduced phagocytic activity, which are involved in inflammation-mediated neurotoxicity. Created in BioRender.com.
Alzheimer’s Disease: Aberrant microglial activation represents a common pathological feature of several neurodegenerative diseases, including AD. Brain amyloid β deposition, a major pathological hallmark of the disease, can induce microglia activation. In this regard, an upregulation of Kv1.3 and Kv1.5 channels was found in Aβ-treated rat microglia and Kv1.3 mediates Aβ-induced microglial ROS production and Aβ-induced microglial priming [93,94]. Of note, Maezawa et al, showed that pro-inflammatory and neurotoxic microglial responses induced by amyloid-β oligomer required Kv1.3 activity in vitro and in hippocampal slices from mouse models of AD pathology [95]. Recent flow cytometric studies identified a distinct pro-inflammatory subset of CNS mononuclear phagocytes (CNS-MPs) that express the gene Kcna3, which encodes the Kv1.3 K+ channel [96]. Interestingly, the expression of Kv1.3 changes during disease, starting with a maximal increase at six months of age followed by a significant decrease between 10 and 12 months of age, a result that is accompanied by published electrophysiological data [95,96]. However, the downregulation of Kv1.3 expression was not observed in human tissue: elevated Kv1.3 channel expression has been observed in human postmortem AD brains by immunohistochemistry and by western blot analysis, and the elevation is limited to microglia, especially in those associated with amyloid plaques, suggesting that Kv1.3 could still represent a relevant microglial target in AD [97]. Furthermore, long-term Kv1.3 blockade in AD mouse models inhibits proinflammatory gene expression in microglia while promoting phagocytic uptake and clearance of Aβ [98]. It is possible that current transgenic models of disease perhaps do not completely replicate microglia phenotypes in human Alzheimer’s disease; further studies are needed to better define these discrepancies [97]. In addition to Kv1.3, also KCa3.1 was reported to play a role in AD. Recent studies demonstrated that the KCa3.1 channel is upregulated in post-mortem sections of AD patients, and in 5XFAD transgenic AD mice starting from three to four months of age, at the insurgence of Aβ amyloidosis and microglial activation. [99]. In this work, the authors also showed that inhibition of KCa3.1 mitigates some AD-like hallmarks including neuroinflammation, enhancing hippocampal neuronal plasticity and amyloid pathology. The same authors previously demonstrated that amyloid-β oligomers (ABO), at low (nanomolar) concentrations, increased microglia nitric oxide (NO) production, causing neuronal damage in vitro, in a KCa3.1-dependent way [100].
Parkinson’s Disease: Many different studies demonstrated that neuroinflammation is critical to PD progression. The most important hallmark of PD is the presence of Lewy bodies, which contain aggregates of α-synuclein [101]. Microglial Kv1.3 is transcriptionally upregulated in response to aggregated α-synuclein (αSynAgg) in primary microglial cultures and animal models of PD, as well as in postmortem human PD brains [102]. Recent evidence demonstrated that PD microglial KCa3.1 also play a critical role in the progression of the disease. KCa3.1 inhibition or gene deletion reduced dopaminergic (DA) neuron loss and improved the locomotor ability reducing microgliosis-mediated neuroinflammatory cytokine production in a mouse model of PD [103].
Amyotrophic Lateral Sclerosis: Several studies described that an inflammatory microenvironment contributes to motor neuron degeneration and disease progression in ALS [104,105], and we have recently shown that CNS infiltrating natural killer cells (NK) contribute to sustain an inflammatory microglia phenotype [21]. In accordance, we have described that in hSOD1G93A, a mouse model of familial ALS (fALS), spinal microglia overexpress KCa3.1, and that the blockade of this channel induces a significant delay in the appearance of motor symptoms [89]. Specifically, a pharmacological blockade with KCa3.1 inhibitor, 1- [(2-chlorophenyl)diphenylmethyl]-1H-pyrazole (TRAM-34), has beneficial effects in rodent models of ALS, reducing the expression of inflammatory factors such as TNF-α, Il-1β, Nos2 and increasing anti-inflammatory factors such as arg1, cd163, sosc3, ym1, bdnf and p2yr12 in the spinal cord. In line with the gene expression data, spinal microglia from TRAM-34-treated mice at the symptomatic stage are less amoeboid, have smaller soma and higher branching complexity, indicating that blockade of KCa3.1 activity might restore the patrolling activity of microglia. Furthermore, TRAM-34 treatment delayed motor symptom appearance in the hSOD1G93A ALS mouse model, as shown by prolonged muscle strength and motor coordination, and increased mice survival. Recently, we found that microglial KCa3.1 is linked to hypothalamic neuroinflammation and affects feeding behaviour in ALS mouse models by restoring homeostatic microglia and attenuating weight loss [106]. It is important to note that a molecule structurally related to TRAM-34, Senicapoc, which has been previously found to be safe in humans in Phase I, II and III clinical trials [107,108] would be available for repurposing, and has been deposited by Pfizer for exactly this purpose in the National Center for Advancing Translational Research (NCATs) library.
Other Pathological Conditions
Ischemia: Post-stroke inflammation plays an important role in brain tissue damage and microglia are the primary immune cells involved in this process, determining the severity of neuroinflammation. Microglia/macrophages acutely isolated from the infarct area in mice with middle cerebral artery occlusion (MCAO), a model for ischemic stroke, expressed Kir2.1, Kv1.3 and KCa3.1 channels [47]. Furthermore, genetic deletion and pharmacological blockade of KCa3.1, using TRAM-34, increased neuronal survival and reduced microglia activation and infarct size [47]. Subsequently, the same authors observed Kv1.3 staining on activated microglia in ischemic infarcts in mice, rats, and humans, and found that Kv1.3-inhibitor PAP-1 reduces secondary inflammatory damage after ischemia/reperfusion [109].
Epilepsy Several anti-inflammatory drugs have documented anticonvulsive effects, indicating the potential role of inflammation to the pathology of epilepsy [110]. Many studies report evidence that microglia are involved in epileptic processes. In particular, microglia seem to play a critical role, releasing increased levels of pro-inflammatory mediators, which may lead to neuronal hyperexcitability and neurodegeneration [110,111,112]. Studies on human microglia isolated from the temporal cerebral cortices of adult epileptic patients showed a higher expression of KCa3.1 in comparison with non-epileptic brains [84,86]. Nevertheless, TRAM-34 treatment, in two different chronic epilepsy models, did not prevent seizures and even exacerbated pathology-related neuronal cell loss [113], thus suggesting that further studies are necessary to understand the effects of KCa3.1 activity in epilepsy. Recently, it has been shown a predominant functional expression of Kv1.3 channels in hippocampal microglia activated after status epilepticus in CX3CR1eGFP/+ mice [114].
Cancer: In Glioblastoma Multiforme (GBM), the most common primary brain tumor in the adults, microglia/macrophages (M/Mφ) represent the major infiltrating cell population. KCa3.1 channels are attractive therapeutic targets for brain tumors, mainly because they are highly expressed in both GBM cells [50] and tumor associated microglia [58], while they are poorly expressed in normal CNS [115]. We have demonstrated that KCa3.1 inhibition by shRNA or by pharmacological tools significantly reduced the tumor-infiltrated cerebral area, and also decreased astrogliosis and M/Mφ activation at the boundary of the tumor, suppressing M/Mφ phagocytosis and migration [116]. We also demonstrated that the KCa3.1 channel blockade reduced tumor invasion and growth with direct effects on glioma, and it switches the activation state of tumor-associated microglia/macrophages (TAMs) towards an antitumor phenotype [78]. In addition, we reported that KCa3.1 activation regulates microglia cell proliferation and neurotoxicity [58]. Kv1.3 is involved in tumor associated astrocyte functions, indeed its inhibition reduced astrogliosis and increased glutamate clearance, reducing excitotoxic neuronal death. In addition, the selective inhibition of the channel reduced tumor growth and directly affected the invasive properties of tumor cells, confirming a key role of Kv1.3 channels as modulators of glial cells [79].
This entry is adapted from the peer-reviewed paper 10.3390/biom11121774
This entry is offline, you can click here to edit this entry!