Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Tocotrienols (T3s), members of the vitamin E family, are natural compounds found in various food sources and exist as four naturally occurring analogues known as alpha (α), beta (β), delta (δ), and gamma (γ).
- tocotrienols
- colorectal cancer
- cell lines
- biomarkers
- Cytoscape
- PRISMA
- KEGG
- STRING
1. Introduction
Colorectal cancer (CRC) is the world’s third most prevalent malignancy and the fourth leading cause of cancer mortality, with almost 1.4 million new cases and approximately 700,000 deaths reported annually [1]. Considering its prevalence, by 2030, the CRC burden is projected to rise by 60%, to over 2.2 million new cases and 1.1 million deaths [2]. More than two thirds of all patients and about 60% of CRC-related deaths are found in countries with a high or extremely high human development index (HDI) [1,2].
Tocotrienols (T3s), members of the vitamin E family, are natural compounds found in various food sources and exist as four naturally occurring analogues known as alpha (α), beta (β), delta (δ), and gamma (γ). The general chemical structure of tocotrienols is shown in Figure 1. Each analogue has different functional groups that are denoted by R1, R2, and R3 [3]. In α-tocotrienol, R1, R2, and R3 represent methyl (Me) groups, but in β-tocotrienol, R1, R2, and R3 represent Me, hydrogen (H), and Me groups, respectively. In γ-tocotrienol, R1 represents H while R2 and R3 represent Me groups. However, in δ-tocotrienol, R1 and R2 represent the H group while R3 stands for the Me group [3]. The T3s are unsaturated and possess an isoprenoid side-chain, which allows them to efficiently penetrate tissues with saturated fatty layers. The anticancer potential of T3s is only beginning to receive recognition as several mechanistic studies have shown that T3s have unique anticancer properties, which are modulated through several cancer-associated mechanisms and pathways (Figure 1) [3,4], such as inhibition of telomerase activity through suppression of protein kinase C (PKC) activity in cancer cells [5]; and blocked expression of hypoxia-induced vascular endothelial growth factor (VEGF), interleukin-8 (IL-8), and cyclooxygenase 2 (COX-2), which play critical roles in cancers as these act as autocrine growth factors for carcinogenesis and tumor neovascularization [6,7,8]. On the contrary, T3s are reported to enhance the expression of p21 and p27, both known to cause cell cycle arrest [9]. In addition, T3s can induce apoptosis in cancer cells through several mechanisms, which includes inhibition of the nuclear factor-κB (NF-κB) pathway and its regulated gene products [10] as well as regulation of both intrinsic and extrinsic apoptotic pathways by modulating the caspase cascades, expression of B cell lymphoma 2 (BCL2), BCL2-associated X protein (BAX), and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) by upregulating the expression of death receptors (DRs) [11,12].
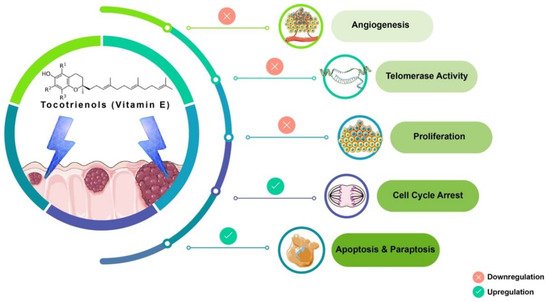
Figure 1. Anticancer actions of tocotrienols. Tocotrienols have been reported to exert anticancer effects by several mechanisms, such as induction of cell death via apoptosis and paraptosis in cancer cells, cell cycle arrest, inhibition of proliferation, suppression of the expression of the human telomerase reverse transcriptase (hTERT) in cancer cells as well as inhibition of angiogenesis. [α-tocotrienol: R1 = Me, R2 = Me, R3 = Me; β-tocotrienol: R1 = Me, R2 = H, R3 = Me; γ-tocotrienol: R1 = H, R2 = Me, R3 = Me; δ-tocotrienol: R1 = H, R2 = H, R3 = Me.
Tocotrienol is a form of vitamin E that is gaining recognition for its immense benefits against various types of cancers and other diseases. The anticancer effects of T3s shown in Figure 1 only represent a small portion of the overall T3-based research. The current state of information on this lesser-known type of vitamin E calls for more investigations to garner more scientific evidence that could further strengthen T3s’ many functions and attributes in various experimental models.
2. Discussion
Inherited CRC is now acknowledged as a significant factor in the development of this disease. Genome sequencing-based diagnosis estimates that one-third of CRC patients appear to have familial colorectal cancer (FCC) as a part of their pathogenesis [39]. The initiation of CRC arises from the epithelial tissue due to the accumulation of genetic modulations in specific oncogenes and tumor suppressor genes (TSGs). In the evolution of sporadic CRC, two primary mechanisms of genomic instability have been identified. The first is chromosomal instability (CIN) caused by a cascade of genomic alterations involving activating oncogenes like KRAS and the inactivation of TSG like p53, DCC/SMAD4, and APC. The second, known as microsatellite instability (MSI), is caused by hypermethylation of the promoters of the DNA mismatch repair genes MLH1 and/or MSH2, as well as secondary mutation of genes with coding microsatellites, such as transforming growth factor receptor II (TGF-RII) and BAX [40,41,42]. In comparison, germline mutations in the defined genes may lead to inherited CRC. Those mutations can be in the tumor suppressor gene APC on chromosome 5q as in familial adenomatous polyposis (FAP) or mutated DNA mismatch repair genes in hereditary non-polyposis colorectal cancer (HNPCC) [43,44]. In this systematic scoping review, differentially expressed candidate proteins in response to γT3 or δT3 treatment were retrieved to identify the molecular mechanisms through which these T3s isoforms modulate anticancer effects.
The 16 CBs (BIRC3, BIRC5, CASP3, CASP8, CASP9, CCND1, CDKN1A, CDKN1B, CTNNB1, JUN, MMP9, MYC, PARP1, RELA, VEGFA, and WNT1) short-listed in this study formed three prominent clusters that were part of the apoptotic, transcriptional misregulation, or cancer progression pathways (Figure 2). All three pathways are interlinked and are crucial with respect to anticancer mechanisms. Hence, it is highly likely that the 16 CBs play a pivotal role in mediating anticancer mechanisms induced in human CRC cell lines exposed to γT3 or δT3. A number of the CBs have been reported to play important roles clinically in the carcinogenesis of patients with CRC (Table 1).
Table 1. Summary of clinically relevant genes and proteins modulated by tocotrienols in human colon cancer cell lines.
| Pathways Involved | CBs Modulation by T3s | Reported Effects in Colon Cancer Patients | Ref. |
|---|---|---|---|
| Apoptosis | Caspase 3 ( ) ) |
● Irradiated CRC cells from patients with lower levels of caspase-3 was associated with poor prognosis | [45] |
| Caspase 8 () |
● Higher prevalence of mutations in the caspase-8 genes in invasive carcinomas; reduce apoptotic activity | [46,47] | |
| Caspase 9 () |
● Expression of the caspase-9 gene downregulated in CRC tissue compared to surrounding normal mucosa | [48] | |
| Transcriptional dysregulation in cancer | CDKN1A (p21) () |
● P21 was downregulated in 50% (371/737) of CRC samples | [49] |
Jun family ( ) ) |
● Higher c-Jun expression observed in human colorectal adenocarcinomas | [50] | |
| c-MYC () |
● CRC patients with higher MYC expression significantly shorter progression-free survival time and overall survival | [51] | |
| RELA (NF-kB/p65) () |
● Reduced RELA expression, resulting in deceased activation of the NF-κB signaling pathway, which inhibited carcinogenesis | [52,53] | |
| Cancer progression | CCND1 () |
● CCND1 gene was detected in tumors from about 50% (54 out of 111) of CRC patients; absent in normal mucosa | [54] |
| VEGFA () |
● Elevated expression of the VEGF family, especially of VEGFA, was reported in CRC patients with LNM | [55] | |
| CTNNB1 () |
● CTNNB1 codes for β-catenin, which supports tumor growth ● A significant link between mutations in CTNNB1 gene and MSI |
[56] |
Downregulat.ed; Upregulated; CB: candidate biomarkers; CRC: colorectal carcinoma; LNM: lymph node metastasis; MSI: microsatellite instability; T3s: tocotrienols. Italics refer to the gene name of the protein.This was evident when these CBs were analyzed using another bioinformatics software, i.e., Cytoscape, which shows the connections or interactions between these biomarkers (Figure 7). In this analysis, 10 of the CBs (CASP3, CASP8, CASP9, CCND1, CDKN1A, CTNNB1, JUN, MYC, RELA, and VEGFA) were found to play critical roles in the protein–protein interactions (PPIs) as there were correlated interactions between these CBs. We observed between 12 and15 engagements marked with either of these CBs, being the protein that exerts an effect (source) or affected by the CBs’ action (target) (Figure 7) [30].
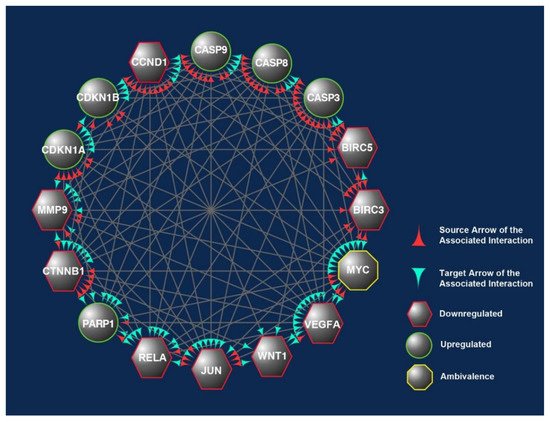
Figure 2. The overlapped targets of the 16 candidate biomarkers (CBs) identified in this study were populated according to their engagements using Cytoscape, an online bioinformatics tool. The protein–protein interactions (PPIs) between these CBs formed a prominent network. The CBs in the hexagons and circles are significantly downregulated or upregulated in human CRC exposed to γT3 or δT3. In contrast, the CB in the octagons is a biomarker that did not show significant changes. The green and red arrowheads reflect the interactions between these CBs. The red arrowheads indicate source CBs protein that can exert stimulatory (circles) or inhibitory (hexagons) or no effects (octagons) on target CBs (green arrowheads).
Caspase-3 (CASP3) is the only CB reported in five independent studies as overexpressed following exposure to T3s. So, therapeutic targeting of caspase-3 may boost cancer cell susceptibility to chemotherapy and irradiation while simultaneously inhibiting invasion and metastasis. Using the CRISPR technology, Zhou et al. [57] established a caspase-3 knockout (KO) human CRC cell line where the caspase gene was knocked out in the HCT116 human CRC cell line (CASP3KO). The authors reported that the CASP3KO-HCT116 cells were less clonogenic, less invasive, and more susceptible to mitomycin-C treatment than the wild-type control cells [57]. In addition, the CASP3KO-HCT116 cells proliferated at a similar rate as the control cells in vivo and were more sensitive to radiation. When administered subcutaneously or intravenously, these cells were less prone to pulmonary metastases than the wild-type HCT116 cells. Deletion of the CASP3 gene also generated lesser EMT phenotypes on a molecular level [57]. In a clinical study, irradiated CRC cells undergoing apoptosis and necrosis were reported to produce significantly higher levels of cleaved caspase-3 (CC3) [45]. The immunohistochemistry staining revealed that the colorectal tumor tissues also showed significantly higher expression of CC3 compared to the peri-tumoral tissues. The authors concluded that high CC3 levels were related to poor prognosis [45]. When the roles of caspase-3 and CC3 were analyzed using the colorectal cancer pathway provided by the KEGG database [58], there was substantial evidence to show that suppression of caspase-3 or increased expression of CC3 suppressed apoptosis, and this could be one of the reasons why this correlates with poor prognosis (Figure 3F).
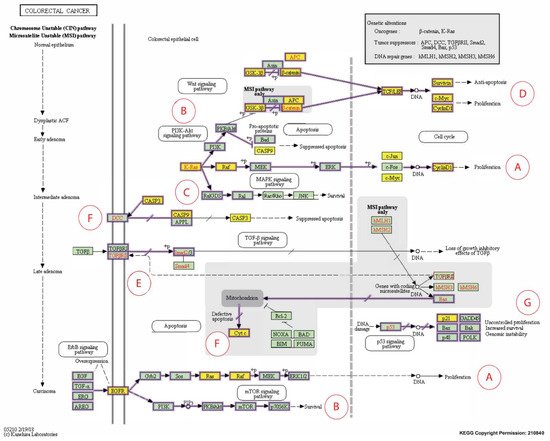

Figure 3. Expanded Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway representation of colorectal carcinoma showing the involvement of the 16 candidate biomarkers selected for this study in the pathogenesis of CRC. The cancer pathways that these 16 CBs are involved in include (A) ERK signaling; (B) PI3K signaling; (C) RAS signaling; (D) WNT signaling; (E) TGFB signaling; (F) Apoptotic signaling; (G) Transcription. Genes marked with yellow rectangles represent the 16 CBs identified in this scoping review.
The CCND1 gene encodes cyclin D1, a biomarker of interest that was reported to be modulated in human CRC cells following exposure to T3s in four out of the 12 short-listed research papers. For instance, in a clinical study, the CCND1 gene was detected in tumors from about 50% (54 out of 111) of CRC patients, but the expression of this gene was absent in normal mucosa [54]. When the cyclin D1 protein expression was investigated in these CRC patients, it was found that this protein could be detected in tumor tissues from 69 cases, of which gene expression was detected in 43 [54]. In the same paper, the authors reported a significant relationship between the expression of the CCND1 gene and protein (cyclin D1). Furthermore, there was a significant link between the expression of the CCND1 gene and metastasis to lymph nodes or distant tissues. Therefore, the author concluded that combined measurement of the CCND1 gene and its protein, cyclin D1, is crucial for use as molecular predictors of human CRC [54]. In addition, when the roles of the cyclin D1 protein were analyzed using the cancer pathway (colorectal cancer) provided by the KEGG database [58], there was substantial evidence to show that downregulation of this protein can reduce the cancer burden (Figure 3A,D) [54,58,59].
Vascular endothelial growth factor (VEGF) is a potent angiogenic protein secreted by almost all types of cancers. The VEGF family includes four ligands and three receptors, of which vascular endothelial growth factor A (VEGFA) is the best-known member. In most papers, exposure to T3s caused downregulation of VEGFA in the CRC cells. Furthermore, in a clinical study involving lymph node metastasis (LNM) from patients with CRC, there was elevated expression of the VEGF family, especially of VEGFA in LNM, which was associated with the patients’ age (p-value < 0.001) [55]. Interestingly, high expression of VEGFA in the primary tumor was positively associated with other ligands and receptors with regards to LNM, implying a mutual effect. Hence, downregulation VEGFA in cancers can be regarded as a good therapeutic outcome.
The apoptosis pathway plays a significant role in inhibiting the tumorigenic progress. A critical factor in the etiopathology of several cancer therapies, such as chemotherapy and irradiation, is to destroy cancers by triggering apoptosis [60]. There are two main apoptotic pathways, i.e., intrinsic and extrinsic pathways. Activation of caspase-8 (CASP8), a cysteine protease, via engagement of various death receptors initiates the extrinsic apoptotic signaling pathway [61,62] and this induces the release of Cyt c from the mitochondria, inducing cell death by activating caspase nine and other apoptosis mediators (Figure 3F).
Five somatic mutations in the CASP8 gene were identified in 98 invasive colorectal carcinomas (5.1%) but not in any adenomas [46]. Of these five mutations, one was the result of a frameshift mutation, one was due to a nonsense mutation, and the remaining three were missense mutations. In addition, the prevalence of caspase-8 mutations was substantially higher in carcinomas (p-value = 0.05) and there was significant reduction of the apoptotic activity in tumors harboring the caspase-8 mutations [46]. It was proposed that the presence of mutant caspase-8 in colon carcinomas showed that mutations in the CASP8 gene may have resulted in the loss of its apoptotic function and restoration of this activity may promote tumor apoptosis for the treatment of CRC [47]. The expression of the caspase-9 (CASP9) gene was downregulated in CRC tissue when compared to the corresponding tissue from normal mucosa (p-value = 0.001) [48]. In addition, patients with downregulated caspase-9 had a lower overall survival (p-value = 0.012) and disease-free survival (p-value = 0.022) [63]. Therefore, caspase-9 (CASP9) could be a useful biomarker in predicting the prognosis of CRC patients [63] (Figure 3F). Exposure to T3s increased the expression of the CASP8 and CASP9 genes in the human CRC cell lines, making it a good target molecule for further studies to evaluate its potential to be used to treat CRC.
CDKN1A (p21) is one of the cyclin-dependent kinase (CDK) inhibitors that is transcriptionally controlled by p53; a transcription factor that plays a vital role in the regulation of the cell cycle. Deletions in the p21 gene were found in 371 (50%) out of the 737 CRC samples analyzed from two prospective cohort studies [49]. Further analysis showed that mutations in a proto-oncogene (BRAF gene) were inversely related to p53 expression and loss of p21 expression. In addition, the correlation between the expression of the p21 gene and mutations in the BRAF gene in the CRC tissues was evident when their p53 status was stratified. In contrast, the relationship between p53 positivity and the mutations in the BRAF genes was no longer evident in CRC when the p21 status was stratified [49]. The relationship between BRAF and p21 genes can be observed in the KEGG colorectal cancer pathway (Figure 3A–C) [58]. Different cancer prognosis and survival types have been linked to somatic changes in genes that regulate cell division. P21 is a crucial regulator of the cell cycle (Figure 3G) [64].
The CTNNB1 genes encode β-catenin protein, which support tumor growth. In 80 human CRC tumor specimens stratified by the presence or absence of microsatellite instability (MSI), mutations in the CTNNB1 gene were found in 53 tumor specimens (25%) with high-frequency MSI (MSI-H) but no mutations were observed in the CTNNB1 gene in the 27 MSI tumors with low-frequency MSI (MSI-L) [56]. The authors concluded that there was a significant link between CTNNB1 mutations and MSI [56]. Furthermore, 46% of the CTNNB1 mutations in endometrial cancer were reported to be immediately phosphorylated by glycogen synthase kinase-3 (GSK-3β). It was proposed that the discrepancies in the mutation profiles show that CTNNB1 mutations may have molecular fingerprints determined by biological factors, such as tumor type and underlying genomic instability pathways, so-called transcriptional misregulation pathways [65]. The proposed role of β-catenin is shown in the KEGG human colorectal cancer pathway (Figure 3D) [58].
The Jun family genes’ products, such as c-Jun, JunB, and JunD, are crucial components of the activating protein-1 transcription factor complexes, which regulate cell proliferation, differentiation, and neoplastic transformation [50]. Although higher c-Jun expression has been observed in many studies concerning CRC (Figure 3A), the expression of JunB and JunD in these tumors has yet to be investigated. Therefore, Wang and his team looked at the expression of c-Jun, JunB, and JunD proteins in 24 cases of human CRC [50]. In identical colectomy specimens, normal-appearing colonic mucosa far from the tumors was employed as a comparison point. According to the findings, in normal mucosa, both c-Jun and JunB proteins were undetectable or barely detectable, but their expression levels were dramatically enhanced in human colorectal adenocarcinomas. JunD protein, on the other hand, was abundant in normal mucosa and only showed a slight increase in adenocarcinomas. These findings point to the possibility that distinct Jun proteins play diverse roles in colonic epithelial cell proliferation and carcinogenesis. Its downregulation might be key to limiting cancer spread.
Epidermal growth factor receptor (EGFR) is a significant oncogene found in various malignancies [66]. Anti-EGFR resistance in metastatic colorectal cancer (MCRC) may be linked to changes in the transcription factor c-MYC (MYC) (Figure 3A,B,D). The expression of MYC was quantified in 121 MCRC patients who had wild-type RAS and BRAF genes before and after treatment with a combination of anti-EGFR and Folfiri therapy as well as in 33 subsequent metastases collected during target therapy [51]. When compared to patients with low MYC expression (LME), those with higher MYC expression (HME) had a significantly shorter progression-free survival time (PFS) and overall survival (OS) [51]. Furthermore, after TT, the HME pattern was substantially more common in metastases, related to anti-EGFR molecular resistance changes. Furthermore, expression gene profiling revealed that MYC plays a critical role in CRC-related cell cycle, apoptosis, signaling, and cell growth pathways. Patients with anti-EGFR-treated MCRC who have higher MYC expression may have a shorter PFS and OS. Identifying specific miRNAs involved in regulating the MYC pathway and downstream MYC effector genes may provide a new target for overcoming anti-EGFR resistance in MCRC. Although few studies suggested that MYC overexpression might sensitize CRC cells to induced apoptosis, we generated a contradicting finding in this review about its up- and downregulation [67,68,69].
RELA/p65, a vital element of the NF-κB signaling cascade, has various roles in oncogenesis. Apart from being an essential member of RNA metabolism, RNA helicase p68 also works as a transcriptional coactivator of numerous oncogenic transcription factors, including β-catenin, and has been linked to cancer progression. Khare et al. found that in both standard and CRC patient samples, the proteins p68, β-catenin, and RELA exhibit a strong positive connection [70].
Both p68 and β-catenin elevated RELA mRNA and protein expression. RELA promoter activity was increased by p68, β-catenin, and Wnt (Figure 3D). p68 and β-catenin knockdown, on the other hand, decreased RELA promoter activity and resulted in lower RELA mRNA and protein expression. p68 was thought to occupy the RELA promoter with β-catenin at the TCF4/LEF binding element (TBE) sites, resulting in RELA transcription being potentiated. The p68 and β-catenin alliance positively regulated the NF-κB target genes. Findings in clinical samples confirmed that p68 increased NF-κB target gene expression. Tumors stably expressing p68 in a mouse transplant model confirmed the in vitro findings. This novel mechanism explains how p68 and β-catenin work together to regulate RELA expression and stimulate the NF-κB signaling axis to promote colon carcinogenesis. This mechanism proposes a potential therapeutic target in CRC by inhibiting NF-κB [52,53]. Integrative proteogenomic profiling appears to have revealed novel therapeutic opportunities for targeting signaling proteins in colon cancer treatment. This unique theory could pave the way for significant progress in molecularly driven precision treatment for colon cancer [71].
This entry is adapted from the peer-reviewed paper 10.3390/nu13114056
This entry is offline, you can click here to edit this entry!