Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Endocrinology & Metabolism
Both in situ and allograft models of cancer in juvenile and adult Drosophila melanogaster fruit flies offer a powerful means for unravelling cancer gene networks and cancer–host interactions.
- tumor–host interactions
- inflammation
- innate antitumor immunity
1. Introduction
Cancer is a major public health issue that causes close to 10 million deaths every year [1]. The growth of a malignant tumor in a specific organ or tissue often induces its dysfunction, but it is mostly the systemic physiopathological alterations that occur in distal organs that result in devastating outcomes and contribute to the death of the host.
These general effects, which were once thought to be mere metastases, are now known to also be a consequence of secreted proteins and hormones, exosomes, and/or metabolites that enter the bloodstream, affecting different organs and provoking physiological alterations that can result in weight loss, anorexia, fatigue, chronic pain, and other debilitating conditions.
2. Immune Responses: Local and Systemic Inflammation
There is a large amount of evidence revealing the dual nature of the immune system, when defining cancer outcome, in either preventing or promoting tumor growth and metastasis [4,5,6]. However, the study of this duality is enormously challenging due to the complexity of the mammalian immune system [7]. Insects rely on an evolutionarily conserved innate immune system, of which studies have served as a blueprint to identify key aspects of mammalian immunity [8]. These discoveries on immunity and the insight provided into disease mechanisms, as well as the new avenues opened for the development of prevention and therapies against diseases, were awarded the Nobel Prize in 2011.
Much of the work on anticancer immunity has focused on adaptive immune responses, such as T-cell activation. However, inflammation and innate antitumor immunity are also important in the emergence of neoplastic cells and in tumor recurrence, particularly in the step from minimal residual disease to active growing recurrence [9]. Inflammation is an innate defensive reaction in response to harmful stimuli (pathogens or injured tissue) and neoplastic cells. Its main function is to eliminate the initial cause of cell injury, clear out necrotic cells and damaged tissues, and initiate tissue repair [10]. While acute inflammation can be beneficial to protect the host from infections or injuries and preneoplastic cells, this immune response must cease when no longer needed such that chronic inflammation is prevented, as this may foster tumor initiation and metastasis in mammals and in Drosophila [11,12].
While the relationship between inflammation and cancer was first exposed in 1863 by Rudolf Virchow [13], the molecular mechanisms by which inflammatory signals help cancer cells to thrive continue to remain a mystery. Virchow hypothesized that cancer originates at sites of chronic inflammation and, nowadays, it is widely assumed that a proinflammatory environment constitutes a risk factor for neoplastic growth in cells that acquire overproliferation capacity [14].
Interestingly, as most Drosophila models are based on the development of tumors in situ, this cancer model can be of great value in deciphering the initiating steps that lead from a few neoplastic cells developing into full-blown tumors, for example, how incipient neoplastic cells resist and/or escape attacks of the innate immune cells, and how innate immune cells sense and recognize tumor cells from normal cells. The innate immunity of insects relies on humoral and cellular immune responses to fight microorganisms (bacteria, viruses, and parasites) or injuries (Figure 1). This innate immunity is composed of different organs and tissues, including the fat body, gut, and blood cells [15].
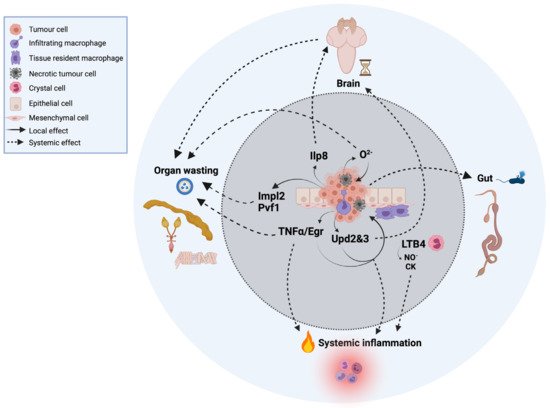
Figure 1. Local and systemic cells and signals in innate immunity and inflammation in cancer in Drosophila. For simplicity, the tumor represented in the image is based on a compilation of different oncogenic signatures in which this review focuses on and is explained throughout the text.
The humoral secretion of antimicrobial peptides (AMPs) from the fat body into the hemolymph has the function of lysing microbes as soon as they reach the epithelial barrier [16]. Some AMPs, specifically drosomycin, have been found to be induced by tumor cells and impact tumor growth [17,18,19].
While mammals have numerous types of immune cells, which participate in the innate and/or adaptive immunity, the cellular immune response of Drosophila involves only three types of circulating blood cells, generally referred to as hemocytes, which are myeloid-like immune cells, including plasmatocytes, crystal cells, and lamellocytes [20,21].
Crystal cells and lamellocytes, which constitute approximately five percent of total immune cells, are required for wound healing and melanization, which is the equivalent to the complement system in mammals [22,23]. The rate-limiting enzymes that mediate the melanization process are the Drosophila prophenoloxidases (PPOs). Crystal cells, in particular, produce PPO1 and PPO2, which contribute to the bulk of melanization in the hemolymph upon injury. Lamellocytes express PPO3, which is believed to contribute to the encapsulation process against parasitoid wasps [24].
Thus far, only plasmatocytes and crystal cells have been implicated in tumor formation and/or tumor-associated inflammation [4]. Plasmatocytes are macrophage-like cells that mediate the phagocytosis of microorganisms and apoptotic cells upon infection or injury. These cells represent ~90–95% of total hemocytes and have the ability to migrate to the tumor site and infiltrate the tumor while producing a cocktail of proinflammatory cytokines [4,25] and chemokines [4,26] (Figure 1). They can be associated with healthy imaginal discs, referred to as tissue-resident macrophages.
Crystal cells play a key role in innate antitumor immunity via the melanization of abnormal and neoplastic cells, and recent studies have uncovered that active crystal cells overexpress the gene CG10602 [26], which encodes a leukotriene A4 hydrolase that catalyzes the synthesis of the eicosanoid lipid leukotriene B4 (LTB4) from arachidonic acid. LTB4 is a proinflammatory mediator that is produced by myeloid cells in mammals [27] in response to an inflammatory insult, and we recently demonstrated its role in tumor-related inflammation in Drosophila Notch-Pten tumors [4], where active lipid proinflammatory mediators are also produced by the tumor cells in Pten-deficient cells.
LTB4 is a potent chemokine and attractor of immune cells [27,28] and pharmacological inhibition or genetic inactivation of 5- and 15-lipoxygenases and leukotriene A4 hydrolase, enzymes that catalyze the synthesis of LTB4, suppress immune cell migration in insects [28,29,30] and tumorigenesis and inflammation in insect larvae cancer models [4,31]. LTB4 also stimulates the production of proinflammatory cytokines and mediators such as nitric oxide (NO) [4,32,33].
Tumor cells communicate locally, promoting an innate immune response in the area widely known as the tumor microenvironment (TME). This local communication can lead to local inflammation.
2.1. Local Proinflammatory Cytokines
One of the most studied cytokines produced by tumors is tumor necrosis factor alpha (TNF-α), whose single homologue is Eiger (Egr) in Drosophila [37,38]. TNF-α/Egr is a major proinflammatory cytokine produced within the TME with a dual role as an anti- and protumoral factor [38], although not all tumors are sensitive to Egr [39]. The proinflammatory role is well-conserved in Drosophila [40], which has permitted us to better understand this duality. This cytokine has both tumor-intrinsic [41,42] and -extrinsic roles, which we discuss below. Thus, the activation of Egr in nontumor cells exerts an antitumoral effect in some Drosophila cancer models, where mutant cells are generated in a wild-type background in imaginal discs. In these contexts, Egr-dependent activation of JNK induces tumor cell death [41,42,43,44]. By contrast, hemocyte-derived Egr has also been shown to promote JNK activation in Drosophila scrib/RasV12 tumors, but in this context, the function of JNK is shifted towards tumor cell proliferation and invasion due to oncogenic cooperation with Rasv12 [45,46]. Interestingly, recent advances in mammals highlight the importance of systemic immunity in response to tumors, although the precise mechanisms remain understudied [47]. The fat body of Drosophila, which functions as a liver and adipose tissue, is the main organ involved in humoral immunity. In 2014, the group of Dr. Marcos Vidal reported that epithelial tumors remotely activate the Toll immune signaling pathway in the fat body to trigger a systemic immune response. They suggest that scrib/RasV12 tumor-derived Egr promotes the production of the Toll ligand Spaetzle (spz) by hemocytes, activating the Toll pathway in the fat body. Toll reciprocally restrains tumor growth by inducing tumor cell death in a non-tissue-autonomous manner [48], but the underlying mechanism by which cell autonomous Toll induces tumor cell death in distant organs remains unknown.
Upds are the Drosophila homologs of mammalian interleukins [49]. Recent work highlights the implication of unpaired 2 and 3 (Upd2 and Upd3) cytokines in local responses to tumors. Upd and Upd2 also act as insect leptin-like cytokines [50], which suggests a pleiotropic effect of these cytokines in local and systemic inflammation. Upd3 is produced by different types of tumors [4,51]. In scrib mutant cells, secreted Upd3 induces JAK/STAT activation in the fat body and hemocytes, which is required for hemocyte proliferation and subsequent tumor suppression [25]. JAK/STAT signaling can exert protumoral effects in different oncogene contexts [25,49,51]. However, in scrib/RasV12 tumors, Upd3 can activate JAK/STAT signal that cooperate with JNK to promote growth and metastasis, whereas in Notch-dependent tumors, JNK is antitumoral [52]. A similar duality exists in mammalian and human cancers [53,54].
2.2. Systemic Inflammation and Metabolism in Cancer
Inflammation and immune responses are often associated with shifts in metabolism, including changes in tumor cells, the host, and immune cells, the latter referred to using the term immunometabolism [55]. Many solid tumors present infiltrating immune cells and release inflammatory cytokines into surrounding tissues and into the bloodstream, which results in systemic inflammation [56].
Systemic inflammation and proinflammatory processes are linked to poor prognosis in patients with cancer, and are often associated with cancer-associated cachexia (CAC), a multifactorial and multiorgan syndrome characterized by a progressive wasting of skeletal muscle and adipose tissue and apparently associated with increased systemic inflammation [57,58]. However, not all cancers with local or systemic inflammation exhibit a wasting phenotype [59]. Nevertheless, CAC is the most-studied whole-body metabolic syndrome associated with cancer and, therefore, it will be the focus of this review in the following sections. This syndrome is not unique to cancer, and several chronic diseases, such as heart failure, infection, obstructive pulmonary disease, and HIV, also lead to cachexia [60]. Advanced-stage cancer patients show CAC 50% of the time, which has an effect on treatment success and patient survival [61], but it can occur even before cancer is first diagnosed.
Cachexia can be accompanied by cancer-associated anorexia, which is not reversed by increasing food intake [58], resulting in significant weight loss, reduced quality of life, and a shortened lifespan [62]. In fact, approximately 30–80% of cancer patients exhibit weight loss depending on the tumor type [63], and up to 30% of people with advanced-stage cancer die not because of the tumor itself, but because of CAC [60,64,65]. Even within the same cancer type, the host physiology and intrinsic differences in the tumor phenotype can lead to variations in the extent to which patients suffer cachexia [66]. In addition, the severity of cachexia is correlated with increased toxicity resulting from chemotherapy which, in turn, provokes further weight loss [60]. Considering the relevance and heterogeneity of such a syndrome, much significant research in the past two decades has focused on CAC, opening many different avenues and providing information, although its underlying mechanisms are still not completely understood.
One of the most important features of cachexia is chronic systemic inflammation, which induces progressive weight and muscle loss [67]. In mammals, it has been shown that different proinflammatory cytokines produced by immune cells and tumor cells can induce cachexia. These include TNF-α, initially termed “cachectin” [68,69], and interleukin-6 (IL-6) [70].
TNF-α has a direct catabolic effect on skeletal muscle through inducing muscle protein degradation [71,72]. In addition, IL-6 is associated with cachexia in rodent models [73,74,75] and is found in high levels in cachectic patients [74,76]. IL-6 induces suppression of protein synthesis in muscle cells [73,74,75] and also induces lipolysis [77]. In Drosophila, it was recently described that JAK/STAT and TNF-α/Egr signaling are elevated in cachectic muscle and promote tissue wasting in a model of scrib/RasV12 tumor-bearing larvae [78], which recapitulates the “high inflammation” that is a hallmark of human cancer cachexia. This study, although it does not demonstrate that TNF-α/Egr is derived from the tumor tissue, constitutes an interesting proof of principle. Here, below, we review the discoveries made in Drosophila that have shed light into the mechanisms of cancer cachexia.
2.2.1. Tumor-Secreted Factors Involved in Cachexia in Juvenile D. melanogaster
Animals have evolved mechanisms to sense and withdraw from physiological and environmental perturbations to maintain stability and homeostasis [79]. In the fly larvae, perturbed growth, injured tissue, and tumor cells activate the Drosophila relaxin peptide Ilp8 (insulin-like peptide 8) [80]. Relaxin peptides belong to the same superfamily as the insulin and insulin-like growth factor (IGF) peptides, but they act through distinct receptors, namely the guanine nucleotide binding protein (G protein)-coupled receptors. Ilp8 is cell-autonomously activated in many if not all tumor cell types in Drosophila that develop from diploid cells. Once produced, Ilp8 is then readily secreted in the hemolymph and activates a developmental checkpoint that delays developmental timing and influences global systemic growth (Figure 1) [80].
Ilp8 binds and activates the relaxin receptor Lgr3 (leucine rich repeat-containing G protein-coupled receptor 3) in the central nervous system (CNS) in a still poorly characterized neural circuit that involves insulin-producing cells, juvenile hormone (JH)-regulating neurons, and the prothoracicotropic hormone (PTTH). Tumor-derived Ilp8 activation of Lgr3 in the brain then inhibits production of Ilp3, JH, and PTTH and, consequently, the production of the maturation hormone ecdysone [80,81,82,83,84].
As such, larvae with tumors induced in the imaginal discs, the brain, or blood cells activate Ilp8 and the developmental time checkpoint, delaying sexual maturation and extending the time imaginal discs spend fostering tumor growth and the cachexia-like fat body waste phenotype (Figure 1).
Ilp8 was independently discovered by two groups in screens to identify candidate genes that mediate tumor-associated developmental delay using oligonucleotide microarrays [80] in the eyeful cancer paradigm [85] and in an unbiased RNAi-based screen [81]. Garelli et al. (2012) also showed that an Ilp8 allele, Ilp8MI00727, serves as a powerful tool in cancer studies. Ilp8MI00727 (an eGFP protein trap line) is silenced in normal growing cells or expressed at low levels but becomes strongly activated in a cell-autonomous manner in response to tumor growth, and this was visualized in vivo by the eGFP protein [80]. As Ilp8 is activated in tumor cells in a nearly universal manner, and in proportion to tumor burden [80,81,86], regardless of the driving oncogene, it serves as a real-time, accurate, in vivo tumor sensor.
2.2.2. Tumor-Secreted Factors Involved in Cachexia in Adult D. melanogaster
The first observations of a wasting phenotype in flies were made by Elisabeth Gateff and Howard A. Schneiderman in 1974. They noticed that flies transplanted with imaginal discs mutant for the tumor suppressor lethal (2) giant larvae (l(2)gl) develop what they called “the bloating syndrome”. The abdomen of these tumor-bearing flies became swollen and translucent, and the fat body and ovaries degenerated [88]. Strikingly, although this phenotype is very robust, the molecular basis remains unknown.
Decades later, Figueroa-Clarevega and Bilder (2015) found that transplanted tumor eye imaginal discs (scrib/RasV12) in adult flies can induce cachexia-like phenotypes [89]. They also identified the tumor-secreted factor imaginal morphogenesis protein-Late 2 (ImpL2), a secreted insulin-signaling antagonist that functions by directly binding to Drosophila insulin-like peptide 2 (Ilp2) [90]. ImpL2 is the fly homologue of the human insulin growth factor binding protein (IGF binding protein) and drives wasting by reducing insulin signaling in peripheral tissues of tumor-bearing adults. However, tumor-specific inhibition of ImpL2 only partially ameliorates the wasting phenotype, indicating the need to uncover other aspects of tumor–host interactions by means of investigating tumor-derived metabolites. Consistent with those findings, the group of Nobert Perrimon [87] showed that ImpL2 is a cachexic mediator in a different fly tumor model overexpressing yorkie (yki) in the adult midgut, which leads to wasting of the ovary, fat body, and muscle associated with systemic insulin resistance [87], a feature also reported in human patients and mouse models of cachexia [91,92]. In addition, these gut tumors also perturb whole-body metabolism by increasing hemolymph trehalose and diminishing glycogen and triglyceride storage. Nevertheless, depletion of ImpL2 in these tumors induces a significant but not total rescue of the organ-wasting phenotypes. These results point out the existence of additional mechanisms contributing to cachexia. Moreover, Perrimon’s lab reported in 2019 that yki-induced gut tumors secrete Pvf1, which triggers host Pvr/MEK signaling and wasting of muscles and the fat body [93], suggesting a role for the MEK/ERK pathway in promoting catabolism in peripheral tissues [94,95].
This entry is adapted from the peer-reviewed paper 10.3390/cells10113211
This entry is offline, you can click here to edit this entry!