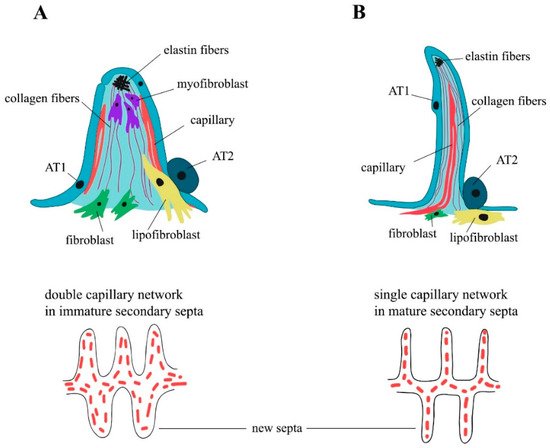
3. Elastogenesis and Collagens in Alveologenesis
ECM plays an important role in lung development, normal functioning, and repair after injury. The main components of the matrix are elastin, fibronectin, collagens I, III, IV, VI, and XVII, laminins, proteoglycans, and glycoproteins. Many diseases that affect the structure and function of the lung are accompanied by the dysregulation of basement membrane components [
107,
108,
109]. While it is well known that the overexpression of collagen fibers causes lung fibrosis, there is not much information on the role of collagens in alveologenesis. Loscertales and colleagues showed that collagen IV is essential in alveolar lung patterning and proposed a model in which collagen IV regulates epithelial and endothelial components important for alveologenesis, induces (in part through the PDGFRα pathway) interstitial αSMA myofibroblasts to proliferate and migrate to the tips of the septa during early septation, and later contributes to the extension and final maturation of secondary septa [
110]. As Col4a1 mRNA is expressed in the interstitium and the tips of alveolar septa in a pattern similar to tropoelastin, the authors suggest that collagen IV regulates elastogenesis specifically in the αSMA
+ cell population. Moreover, Col4a1 mutants have an increased number of cells with lipid content (AT2 and lipofibroblasts) and abnormal capillary formation. An interesting observation is that collagen VI promotes lung epithelial cell spreading and increases the rate of “wound healing” in response to a scratch injury via phosphoinositide 3-kinase (PI3k) and the cell division control 42 homolog (CDC42) downstream of the interaction with b1 integrins [
111].
Among the lung ECM components, collagen fibers (mainly type I and III), which are stiff and strong, provide structural integrity and strength to the lung and are the most important load-bearing element within the alveolar duct and wall, while elastic fibers provide tissue resilience [
112]. Degradation of elastic fibers by elastases (e.g., smoking induces the secretion of elastases from inflammatory cells, and mice deficient in macrophage elastase were resistant to tobacco smoking that normally provokes emphysema in wild-type mice, which was shown by [
113]) or aging give rise to emphysema. The formation of functional elastin fibers, which give elasticity to the alveoli, plays a crucial role in the process of alveologenesis. Tropoelastin, a soluble monomer of mature elastin, is a spring-like molecule that is extremely flexible and extensible [
114]. In this manner, elastin itself possesses remarkable durability and thus the ability to undergo many stretch–recoil cycles while maintaining the structural and functional integrity of elastic tissues throughout life. Elastogenesis is a highly hierarchical multistep process [
115]. Elastin and fibrillin-1 (FBN1) represent the building blocks of elastic fibers that compose the elastic fiber core and microfibrils, respectively [
116]. The formation of elastic fibers requires several accessory proteins, including fibulin-4, fibulin-5, and the latent TGF-beta binding protein, LTBP-4, which shares structural homology with FBN1 [
117,
118].
Elastin is expressed in the lung by several types of cells, including pleural mesothelial cells, airway and blood vessel smooth muscle cells, endothelial cells, and interstitial fibroblasts. Due to this, a system of elastic fibers is formed, which ensures an equal transfer of the applied force to all parts of the lung [
91]. Numerous studies show that the formation of alveoli is closely related to the formation of elastin fibers in the respiratory region of the lung. During septation, the formation of elastin fibers reaches its peak. Three phases of tropoelastin expression have been observed in the developing lung, with each phase characterized by a distinct expression pattern [
50]. In the first phase, on E15.5, the expression of tropoelastin mRNA was confined to mesenchymal cells lining the proximal tubules of the endoderm. During the second phase of expression, at the saccular stage on E18.5, almost all lung mesenchyme was tropoelastin-positive. It was also expressed in the developing vascular walls. In the third phase, on P7–P14, tropoelastin expression was limited to a population of mesenchymal cells located on the tips of the growing alveolar septa (and to developing cells of the vascular and bronchial walls). Mesenchymal tropoelastin-positive cells detected in the third phase of postnatal expression were specifically and completely absent in the lung of PDGF-A
−/− mice, while tropoelastin expression in bronchial and blood vessel walls was unaffected. The impairment of postnatal alveologenesis in PDGF-A
−/− mice was suggested to be due to a prenatal block in the distal distribution of PDGFRα
+ cells [
50]. In the absence of a PDGF-A signal, PDGFRα
+ cells are unable to multiply and spread. Thus, the depletion of myofibroblasts in PDGF-A-null mice results in a lack of elastic fibers in the alveolar walls and incomplete alveolarization [
27,
50]. Similar effects were observed in Hoxa5
−/− murine lung, in which the subset of the mesenchymal progenitors of alveolar αSMA
+ myofibroblasts expressing Pdgfrα failed to spread into the distal fetal lung [
152]. They were trapped in the parenchyma surrounding future alveoli, where they produced abnormal elastic fibers. Elastic fibers were disorganized and aberrantly distributed within the pulmonary tissue on P0 and P5, and alveologenesis was impaired. From P15 onwards, the fibers appeared more abundant, disorganized, and fragmented. Northern blot analyses of tropoelastin expression failed to reveal differences in tropoelastin transcript levels between wild-type and Hoxa5
−/− lungs. Later, it was shown that Hox5 genes are required in elastin network formation by regulating the adherence of the alveolar fibroblasts to fibronectin, which is disrupted because of the loss of the integrin heterodimer Itga5b1 in mutant fibroblasts [
153]. In Hoxa5 mutants, the activated macrophages were abundantly found in the lung as early as birth, and most of them expressed matrix metalloproteinase 12 (MMP12). This recruitment coincided with the abnormal elastin deposition by the mispositioned alveolar myofibroblasts, supporting the notion that elastin fragments are chemotactic for monocytes [
154]. Furthermore, recent studies showed that the absence of Pdgfra in Gli1
+ SCMFs disrupted the expression of elastogenic genes, including members of the LOX, fibronectin, and fibulin families, while the expression of the EGF family members was increased, and Tgfb1 was suppressed in the lung fibroblasts of the studied mice. Similar results were found in lung samples from a person with BPD. Blocking PDGF-A signaling in vitro had the same effect. In addition, the effect was reversible by inhibiting EGF or activating TGF-beta signaling. These data demonstrate the role of PDGFA/PDGFRα in the control of elastogenic gene expression through the secondary level of signaling networks consisting of EGF and TGF-beta [
80]. Finally, the growth factor FGF-18 was also shown to affect myofibroblast differentiation by stimulating the expression of tropoelastin, LOX, and fibulins 1 and 5 during secondary septation [
51].
Some methods that suppress alveolarization, such as prolonged hyperoxia (BPD modeling), also suppress septation and elastin expression in the alveolar wall [
155,
156,
157,
158]. Elastin fiber assembly impairment in newborn rats due to copper deficiency [
159] or the administration of beta-Aminopropionitrile (beta APN) [
160], which interferes with the synthesis of elastin and collagen by inhibiting LOX, results in the impairment of alveolar septa formation and the simplification of the alveoli by the type of emphysema. In contrast to PDGF-A
−/− mice, in which the absence of alveolar elastin synthesis and the abnormal structure of the alveoli are detected after P4, the inactivation of the elastin gene in Eln
−/− mice leads to a delay in the perinatal development of the terminal airway branches. At birth, these mice have enlarged cavities in the lung that resemble emphysema [
161]. These differences indicate that, in addition to its role in the formation of the structure of alveoli, elastin is important for the terminal branching of the airways in lung development. This also supports the idea that elastin is expressed by airway smooth muscle cells, which enwrap the branching tips and the stalk of the primordial airways and physically support branching morphogenesis [
162,
163].