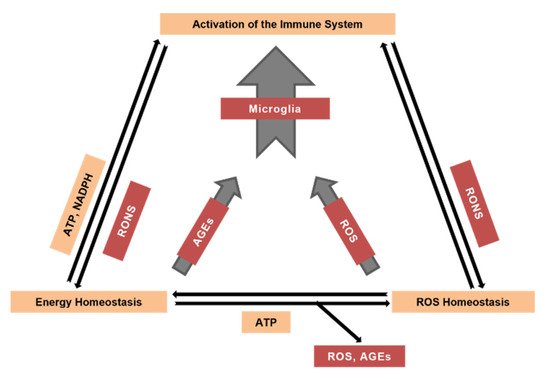
Reactive oxygen and nitrogen species are crucial contributors to the age-dependent decline in all tissues. Neural tissue, one of the main oxygen consumers in the mammalian body, is especially prone to reactive species-mediated damage. Brain cells, including neurons, astrocytes, and microglia, produce reactive oxygen species (ROS) by specific enzymatic systems, including complexes of the mitochondrial respiratory chain, multienzyme flavin-containing complexes, monoamine and xanthine oxidases, microglial and endothelial NADPH oxidases and cyclooxygenases in addition to non-enzymatic and potentially uncontrolled mechanisms of ROS production, such as autooxidation of quinones or other aromatic compounds. Nitric oxide produced by nitric oxide synthases powers the conversion of ROS into reactive nitrogen species (RNS). Both ROS and RNS play important signaling roles and are also capable of modifying other molecules such as proteins, nucleic and fatty acids, lipids and carbohydrates. The antioxidant system, comprising low molecular mass antioxidants (e.g., tocopherol, ascorbic acid and glutathione) and high molecular mass antioxidants such as enzymes (e.g., catalases, peroxidases, superoxide dismutases) and others, protects cells from potential damage caused by ROS or RNS. Powering antioxidant systems by NADPH provides neural tissue with defense against ROS but may also trigger ROS production by NADPH oxidases and cyclooxygenases. In turn, mitochondria start using ketone bodies as an energy source under certain conditions. Increased steady-state levels of ROS and RNS, along with the aforementioned ROS-modified molecules, activate the organisms’ immune system including brain’s microglia.
- Reactive Oxygen Species
- Reactive Nitrogen Species
- Oxidative Stress
- Advanced Glycation End Products
- Brain Aging
- Microglia
1. Introduction
2. Mechanisms of ROS Generation
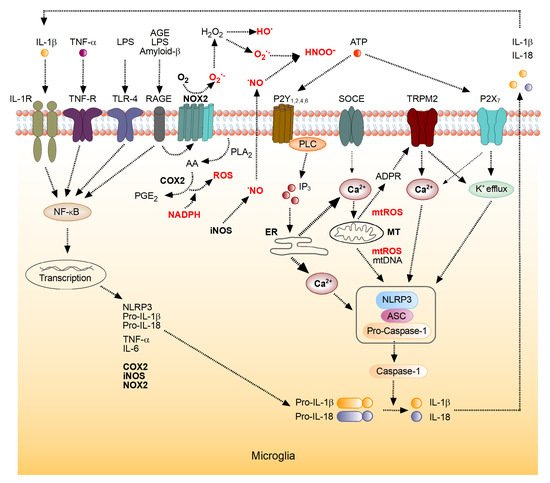
3. The Role of the Brain’s Immune System in the Generation of ROS
This entry is adapted from the peer-reviewed paper 10.3390/antiox10111715
References
- Adam-Vizi, V. Production of Reactive Oxygen Species in Brain Mitochondria: Contribution by Electron Transport Chain and Non-Electron Transport Chain Sources. Antioxid. Redox Signal. 2005, 7, 1140–1149.
- Lushchak, V.I. Free Radicals, Reactive Oxygen Species, Oxidative Stresses and their Classifications. UKR. Biochem. J. 2015, 87, 11–18.
- Garaschuk, O.; Semchyshyn, H.M.; Lushchak, V.I. Healthy Brain Aging: Interplay between Reactive Species, Inflammation and Energy Supply. Ageing Res. Rev. 2018, 43, 26–45.
- Lushchak, V.I. Interplay between Bioenergetics and Oxidative Stress at Normal Brain Aging. Aging as a Result of Increasing Disbalance in the System Oxidative Stress-Energy Provision. Pflugers Arch. 2021, 473, 713–722.
- Semchyshyn, H. Is Carbonyl/AGE/RAGE Stress a Hallmark of the Brain Aging? Pflugers Arch. 2021, 473, 723–734.
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950.
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300.
- McCord, J.M.; Fridovich, I. Superoxide Dismutase. An Enzymic Function for Erythrocuprein (Hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055.
- Reczek, C.R.; Chandel, N.S. ROS-Dependent Signal Transduction. Curr. Opin. Cell Biol. 2015, 33, 8–13.
- Zandalinas, S.I.; Mittler, R. ROS-Induced ROS Release in Plant and Animal Cells. Free Radic. Biol. Med. 2018, 122, 21–27.
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383.
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743.
- Longo, V.D.; Di Tano, M.; Mattson, M.P.; Guidi, N. Intermittent and Periodic Fasting, Longevity and Disease. Nat. Aging 2021, 1, 47–59.
- García-Revilla, J.; Alonso-Bellido, I.M.; Burguillos, M.A.; Herrera, A.J.; Espinosa-Oliva, A.M.; Ruiz, R.; Cruz-Hernández, L.; García-Domínguez, I.; Roca-Ceballos, M.A.; Santiago, M.; et al. Reformulating Pro-Oxidant Microglia in Neurodegeneration. J. Clin. Med. 2019, 8, 1719.
- Cepas, V.; Collino, M.; Mayo, J.C.; Sainz, R.M. Redox Signaling and Advanced Glycation Endproducts (AGEs) in Diet-Related Diseases. Antioxidants 2020, 9, 142.
- Garaschuk, O. The Role of NLRP3 Inflammasome for Microglial Response to Peripheral Inflammation. Neural Regen. Res. 2021, 16, 294–295.
- Tschopp, J.; Schroder, K. NLRP3 Inflammasome Activation: The Convergence of Multiple Signalling Pathways on ROS Production? Nat. Rev. Immunol. 2010, 10, 210–215.
- Yang, S.; Qin, C.; Hu, Z.-W.; Zhou, L.-Q.; Yu, H.-H.; Chen, M.; Bosco, D.B.; Wang, W.; Wu, L.-J.; Tian, D.-S. Microglia Reprogram Metabolic Profiles for Phenotype and Function Changes in Central Nervous System. Neurobiol. Dis. 2021, 152, 105290.
- Metcalfe, N.B.; Alonso-Alvarez, C. Oxidative Stress as a Life-History Constraint: The Role of Reactive Oxygen Species in Shaping Phenotypes from Conception to Death: Oxidative Stress as a Life-History Constraint. Funct. Ecol. 2010, 24, 984–996.
- Sikora, E.; Bielak-Zmijewska, A.; Dudkowska, M.; Krzystyniak, A.; Mosieniak, G.; Wesierska, M.; Wlodarczyk, J. Cellular Senescence in Brain Aging. Front. Aging Neurosci. 2021, 13, 646924.
- Papa, S.; Skulachev, V.P. Reactive Oxygen Species, Mitochondria, Apoptosis and Aging. Mol. Cell. Biochem. 1997, 174, 305–319.
- Büeler, H. Mitochondrial and Autophagic Regulation of Adult Neurogenesis in the Healthy and Diseased Brain. Int. J. Mol. Sci. 2021, 22, 3342.
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in Neuroplasticity and Neurological Disorders. Neuron 2008, 60, 748–766.
- Fan, L.M.; Geng, L.; Cahill-Smith, S.; Liu, F.; Douglas, G.; Mckenzie, C.-A.; Smith, C.; Brooks, G.; Channon, K.M.; Li, J.-M. Nox2 Contributes to Age-Related Oxidative Damage to Neurons and the Cerebral Vasculature. J. Clin. Investig. 2019, 129, 3374–3386.
- Vignais, P.V. The Superoxide-Generating NADPH Oxidase: Structural Aspects and Activation Mechanism. Cell. Mol. Life Sci. CMLS 2002, 59, 1428–1459.
- Kontos, H.A.; George, E. Brown Memorial Lecture. Oxygen Radicals in Cerebral Vascular Injury. Circ. Res. 1985, 57, 508–516.
- Kukreja, R.C.; Kontos, H.A.; Hess, M.L.; Ellis, E.F. PGH Synthase and Lipoxygenase Generate Superoxide in the Presence of NADH or NADPH. Circ. Res. 1986, 59, 612–619.
- Terada, L.S.; Willingham, I.R.; Rosandich, M.E.; Leff, J.A.; Kindt, G.W.; Repine, J.E. Generation of Superoxide Anion by Brain Endothelial Cell Xanthine Oxidase. J. Cell. Physiol. 1991, 148, 191–196.
- Minaev, B.F. How Cofactor-Free Oxygenases Can Overcome Spin Prohibition in Substrates Oxygenation by Dioxygen. Chem. Phys. 2019, 521, 61–68.
- Hanschmann, E.-M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, Glutaredoxins, and Peroxiredoxins—Molecular Mechanisms and Health Significance: From Cofactors to Antioxidants to Redox Signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605.
- Jones, D.P. Redox Theory of Aging. Redox Biol. 2015, 5, 71–79.
- Asiimwe, N.; Yeo, S.G.; Kim, M.-S.; Jung, J.; Jeong, N.Y. Nitric Oxide: Exploring the Contextual Link with Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2016, 2016, 7205747.
- Brawek, B.; Garaschuk, O. Microglial Calcium Signaling in the Adult, Aged and Diseased Brain. Cell Calcium 2013, 53, 159–169.
- Brawek, B.; Garaschuk, O. Monitoring In Vivo Function of Cortical Microglia. Cell Calcium 2017, 64, 109–117.
- Garaschuk, O.; Verkhratsky, A. Physiology of Microglia. Methods Mol. Biol. 2019, 2034, 27–40.
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S. Calcium, ATP, and ROS: A Mitochondrial Love-Hate Triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833.
- Wang, L.; Negro, R.; Wu, H. TRPM2, Linking Oxidative Stress and Ca2+ Permeation to NLRP3 Inflammasome Activation. Curr. Opin. Immunol. 2020, 62, 131–135.