The intrinsic cellular heterogeneity and molecular complexity of the mammalian nervous system relies substantially on the dynamic nature and spatiotemporal patterning of gene expression. These features of gene expression are achieved in part through mechanisms involving various epigenetic processes such as DNA methylation, post-translational histone modifications, and non-coding RNA activity, amongst others. In concert, another regulatory layer by which RNA bases and sugar residues are chemically modified enhances neuronal transcriptome complexity. Similar RNA modifications in other systems collectively constitute the cellular epitranscriptome that integrates and impacts various physiological processes. The epitranscriptome is dynamic and is reshaped constantly to regulate vital processes such as development, differentiation and stress responses. Perturbations of the epitranscriptome can lead to various pathogenic conditions, including cancer, cardiovascular abnormalities and neurological diseases. These RNA modifications modulate the stability, transport and, most importantly, translation of RNA.
- RNA modifications
- RNA metabolism
- brain development
- neurodegenerative diseases
- neurodevelopmental disorders
1. Introduction
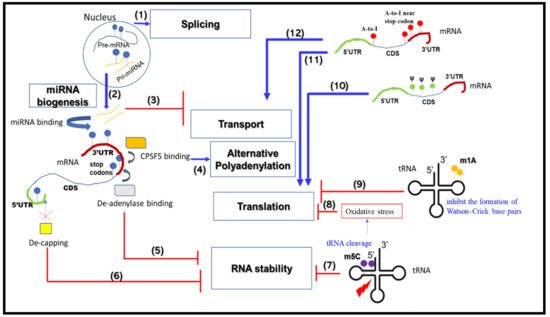
2. RNA Metabolism-Associated Neurological Disease Mechanisms
2.1. mRNA Splicing
2.2. mRNA Alternative Polyadenylation
2.3. mRNA Transport and Translation
2.4. mRNA Stability
2.5. miRNA Biogenesis
|
Disease Type |
Altered RNA Metabolism Pathway |
RBP(s) Involved |
Mechanisms |
Neurological Disease(s) |
References |
|---|---|---|---|---|---|
|
Neuro developmental diseases |
Splicing, Translation |
CPEB4 |
Missplicing of CPEB4 causes reduced inclusion of a neuron-specific microexon, leading to diminished expression of the Cpeb4 transcript that activates translation of mRNAs via polyadenylation under normal conditions |
ASD |
[99] |
|
Splicing Translation, mRNA stability, miRNA biogenesis |
RBFOX1, RBFOX2 (RBM9), RBFOX3 (Neun) |
RBFOX1 binds to the 3′-UTR of its target mRNAs and regulates:
RBFOX2 and RBFOX3 (repression)
Altered splicing of RBFOX family proteins impairs their control of gene expression |
ASD |
||
|
Transport, Translation |
FMRP |
CGG repeat expansion beyond 200 (>200) at the 5′-UTR of FMR1 affects protein expression, resulting in dysregulated spatio-temporal transport/translation of dendritic mRNAs |
FXS |
[64] |
|
|
APA |
NUDT21 |
Elevated amount of NUDT21, a subunit of pre-mRNA cleavage factor Im, due to copy number variation causes abnormal usage of polyadenylation sites, resulting in the generation of an inefficiently translated long isoform of MeCP2 protein. |
Neuropsychia tric disease |
[44] |
|
|
Neuro degenerative diseases |
Splicing |
PRPF8 |
Mutated Huntingtin (HTT) traps PRPF8 (a splicing factor) to cause CREB1 mis-splicing |
HD |
[18] |
|
Translation |
HTT |
Mutant HTT stalls ribosomes |
HD |
[72] |
|
|
Splicing |
MBNL family proteins |
RNA corresponding to expanded microsatellite repeats in DMPK traps MBNL-family proteins, impairing their normal function in splicing |
DM |
[103] |
|
|
Translation |
ATAXIN-2 |
CAG expansion in the reading frame of ATAXIN-2 causes loss of protein function that, under normal conditions, acts as an mRNA translation activator via polyadenylation |
SCA2, ALS |
[75] |
|
|
RAN Translation, Abnormal RNA structure (RNA foci) |
Matrin-3 |
GGGGCC repeat expansion mutation in the C9orf72 gene causes sequestration of Matrin-3 at the RNA foci and RAN translated peptides and loss of function of Matrin-3 |
FTLD, ALS |
[104] |
|
|
mRNA stability, Splicing, Translation |
nELAVL |
nELAVL regulates disease-specific splicing of the pre-mRNAs Picalm and Bin1 by incorporating exons 13 and 6a, respectively. The proteins corresponding to these spliced isoforms have been implicated in trafficking of amyloid precursor protein |
AD |
[105] |
|
|
Transport, Translation, miRNA biogenesis |
TDP-43 |
|
FTLD, ALS |
||
|
Transport, Translation |
FUS |
Normal FUS functions such as axonal transport/translation of mRNAs are adversely impacted in diseased neurons. Under disease conditions, the altered intracellular localization of FUS disrupts its functions as an RBP |
FTLD, ALS |
[109] |
|
|
Splicing, miRNA biogenesis |
hnRNPs, MBNL1 |
mRNA corresponding to shorter CGG repeat expansions (<200) in the 5′UTR of FMR1 sequester many RBPs, e.g., hnRNPs and MBNL1 |
Fragile X-associated tremor/ataxia syndrome (FXTAS) |
[110] |
|
|
APA |
α-synuclein |
Presence of an extended 3′-UTR region in α-synuclein transcript impacts accumulation of α-synuclein protein that is redirected away from synaptic terminals towards mitochondria |
PD |
[48] |
UTR—untranslated region; hnRNPs—heterogenous nuclear ribonuleoproteins.
2.6. Roles for RBPs in RNA Metabolism and Neurological Diseases
3. RNA Modifications that Change RNA Metabolic Processes
This entry is adapted from the peer-reviewed paper 10.3390/ijms222111870