Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Surgery
|
Infectious Diseases
In the past 30 years, diabetic foot infections (DFI) have become increasingly prevalent due to the rising incidence of diabetes mellitus (DM). Twenty percent of diabetes-related hospital admissions in the U.S. are from DFI, which is typically introduced by direct inoculation through a traumatic entry site in an insensate foot.
- diabetic foot infection
- Pathophysiology
- Clinical Assessment
1. Diabetic Susceptibility to Diabetic Foot Infections (DFI)
Several pathological factors place diabetic patients at increased risk for foot infections, including diabetic neuropathy, vascular insufficiency, and immunological dysfunction [12]. A strong link has been reported between diabetic neuropathy and foot ulceration [13]. Diabetic neuropathy is a complex polyneuropathy consisting of peripheral, motor, and autonomic neuronal damage [14].
Peripheral neuropathy is prevalent in 10.9–32.7% of diabetic patients in the U.S. [15,16]. Minor foot trauma from sources such as ill-fitting shoes or injury goes unnoticed from lack of sensation [17]. When repeated or left unattended, this trauma may lead to ulceration (Figure 1) and subsequent infection. Muscle atrophy, foot deformity (Charcot arthropathy, claw foot, pes cavus, hallux valgus), and gait abnormalities are caused by motor neuropathy resulting from the loss of myelinated fibers [18]. These biomechanical changes predispose neuropathic patients to foot ulceration due to increased pressure and shearing. Additionally, a study by Lung et al. suggests that a moderate-to-fast walking intensity decreases plantar stiffness and increases risk of foot ulceration compared to walking at slower speeds [19]. Charcot arthropathy develops in 13% of patients with neuropathy. It can result in fractures, dislocations, and fracture dislocations causing profound deformity and a rocker-bottom appearance of the plantar foot, due to bone breakdown and joint collapse [20,21]. Autonomic neuropathy in the lower limb causes vasodilation and excess warmth in the foot. Moreover, impaired neuronal control of the sweat glands reduces perspiration (anhidrosis), leading to dry skin that is prone to fissure or callus and increasing the risk of developing a foot wound [22,23]. Diabetic patients may also experience vascular insufficiency (micro and macrovascular) and immunological dysfunction, which further predisposes them to ulceration, impaired healing, and infection (Figure 1).
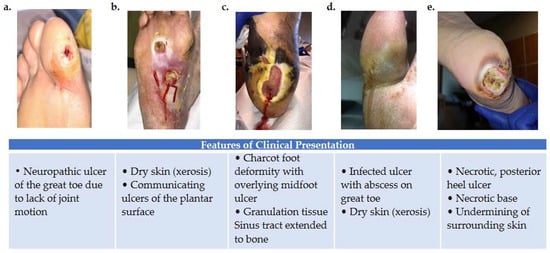
Figure 1. Variations in foot wound location, presentation, and severity as illustrated by images (a–e). (a) Patient presenting with neuropathic ulcer under the proximal interphalangeal joint of the hallux. The ulcer is related to lack of joint motion at the first metatarsophalangeal joint. (b) Patient presenting with an infected ulcer following the flexor tendons of the foot. Notice a blow lesion at the plantar arch. Dry skin (xerosis) is a sign of autonomic neuropathy. (c) Patient presenting with Charcot foot deformity and overlying midfoot ulcer. Macerated skin around the edges of the ulcer and a sinus tract that extends to the bone is also seen. (d) Patient with an infected ulcer with abscess on the great toe and xerosis suggesting autonomic neuropathy. (e) Patient with a posterior heel ulcer containing a necrotic base and undermining of surrounding skin.
2. Clinical Diagnostic Tests
Several diagnostic tests are available to help with the clinical assessment of diabetic foot infections (DFI), including the probe-to-bone test, biopsy, and assessment of inflammatory markers (ESR and CRP). The probe-to-bone test is routinely used during physical examination of the patient to evaluate the potential for OM [24]. In this procedure, a sterile tip probe is introduced through the ulcer to determine if bone is palpable. A solid/gritty end-point is considered positive for OM [25]. Suspected infections may be evaluated for microorganisms by sampling the wound site with an ulcer swab, soft tissue biopsy, or bone biopsy.
Many have argued that the results of these tests should not be independently used for diagnosis and should be only used as a screening method [26]. A negative test result in any of these cases does not exclude the diagnosis of DFI and a positive result may be misleading. For instance, soft tissue biopsies or swab cultures only assess the superficial flora and may not reflect the full extent of infection [27]. Bone biopsies are also limited because the technique requires trained personnel and it is prone to sampling errors. Moreover, there is no standardized definition of a positive bone biopsy and many classifications exist, each with its own merits and weaknesses—further convoluting the problem [27,28]. Historically, classification systems did not include the presence of OM as an indication of infection severity, and it is well known that the presence of OM negatively impacts the outcomes of diabetic foot infections [29]. Without a universal classification system, determinants of infection severity may vary, leading to a diagnostic disagreement among physicians. The accuracy of tests such as the probe-to-bone test relies on the prevalence of OM in the patient cohort being examined. For example, a group of hospitalized patients with severe infection may have a high positive predicative value using these methods, while a group of outpatients with low likelihood of OM may have a low positive predicative value [30]. These factors, as well as others, have encouraged advancements in understanding of the molecular mechanisms in DFI.
3. Molecular Mechanisms of DFI Features
All of the currently available diagnostic tests rely on the assessment of changes in DFI at a tissue level (Figure 2); however, such changes are only detectable when damage has already been inflicted. A strong understanding of the molecular changes that precede visual tissue damage is required to advance the diagnosis and treatment of DFI.
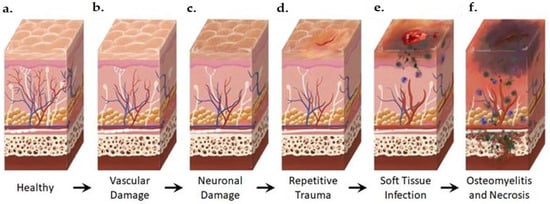
Figure 2. Anatomical Changes Associated with Diabetic Foot Pathophysiology. Diabetic neuropathy is a multi-faceted polyneuropathy related to an increased risk of ulceration, infection, and amputation. Sustained hyperglycemia damages the endothelial lining of the blood vessels in previously healthy tissue (a), leading to impaired circulation (b). Without sufficient vascular support, nerves die off and the skin may become dry and cracked as sweat secretions decrease (c). In the event of injury, numbness in the foot due to neuronal ischemia may mean that insults go undetected for some time (d). Fissures in the dried skin can harbor microorganisms, increasing the likelihood of wound infection. Initial microbial invasion of the trauma site leads to inflammation, vasodilation, and soft tissue necrosis (e). Decreased vascularization compromises immune response to infection and prolongs healing time. If the infection persists, usually because of delayed care or ineffective treatment, microbes may invade bone tissue, leading to osteomyelitis and bone deformation (f). White: neurons, red: arteries, blue: veins, purple: polymorphonuclear lymphocytes, green: microorganisms.
Bone deformities in the foot such as Charcot arthropathy occur when bone destruction from osteoclastic resorption exceeds bone building from osteoblastic recruitment. This breakdown, radiographically visualized as osteolysis, is mediated through the inflammatory RANKL (receptor activator of the nuclear factor-kappa B ligand) pathway and has been identified as an important pathway in the pathogenesis of Charcot foot. When RANKL concentration is higher than its competitive antagonist, OPG (osteoprotegerin), osteoclastogenesis is stimulated, affecting increased osteoclast activity, osteolysis, and deformity [31,32,33].
Arterial atherosclerosis, excessive leukocyte adhesion, increased vascular permeability, impaired hemostasis, and altered proliferation and apoptosis of vascular cells are all factors contributing to diabetic vascular insufficiency [34]. Poorly controlled glucose levels activate the polyol glucose metabolism pathway in which glucose is reduced to sorbitol and subsequently oxidized to fructose. Excess fructose and sorbitol lead to increased osmotic molality and ultimately osmotic stress [35]. Competitive consumption of NADPH and decreased nitric oxide synthetase activity also contribute to this challenge, as low nitric oxide concentration stimulates vessel constriction [36].
In addition to diminished perfusion, immune dysfunction has also been reported [37]. Diabetic patients frequently present with defective neutrophil function irrespective of glycemic status, including disrupted migration patterns associated with reduced production of chemotactic factors, amplified generation of reactive oxygen species, and abated phagocytosis from complement system dysfunction. Collectively, these changes facilitate pathogen invasion and infection development [38]. One retrospective clinical study demonstrated that the ratio of lymphocytes to neutrophils, platelets, and monocytes could predict the need for amputation due to DFI [39].
The innate immune system is a major source of cytokines. Macrophages and dendritic cells may produce cytokines in response to damage-associated molecular patterns (DAMPS) through the activation of pattern recognition receptors (PRRs). These inflammatory cytokines (such as TNFα, IL-1, IL-6, and interferon-ϒ) trigger osteoblast upregulation of RANKL, facilitating osteoclastogenesis [40]. Mitochondrial antiviral signal proteins (MAVS) also stimulate infected cells to secrete cytokines, activating the NF-κB and IRF3 pathways, which regulate the expression of type-I interferons. When bound to type-1 interferon receptors, these cytokines activate the JAK-STAT pathway, inhibiting pathogen replication and assembly due to a heightened expression of interferon-stimulated genes [41]. Type-I interferons such as INF-β are expressed in stromal cells, which are capable of differentiating into mature osteoblasts [42].
The combination of diabetic neuropathy, impaired perfusion, neutrophil dysfunction, and cytokine imbalance encourages infection development and progression, ischemic ulcers, or gangrene, which may culminate in amputation [22]. To avoid amputation and disease progression, a deeper understanding of pathogen involvement is necessary.
A wide variety of microorganisms may cause DFI, the most common of which is Staphylococcus aureus. The methicillin-resistant Staphylococcus aureus strain (MRSA) occurs in 16.78–30% of DFI cases, although this value varies geographically [43,44]. MRSA infection has been shown to have no impact on mortality, but is correlated with an increased rate of hospitalization and higher risk of limb amputation [45]. Amputation is effective in preventing the spread of infection, and studies in the United Kingdom and Germany have found that the procedure increased the life expectancy by 2 years in 50% of the diabetic subjects studied [46,47]. Even so, only 56% of diabetic patients with ulcerative infections were found to survive 5 years after initial onset of the ulcer [47]. Altogether, this information highlights the need for improved ulcer prevention and the prompt diagnosis of DFI. Imaging may define the precise location of the infected bone and establish the border for amputation. Advanced imaging methods may allow for early detection and verify the adequate response to therapy to avoid amputation.
This entry is adapted from the peer-reviewed paper 10.3390/ijms222111552
This entry is offline, you can click here to edit this entry!