Glioblastoma is an immunologically ‘cold’ tumor, which are characterized by absent or minimal numbers of tumor-infiltrating lymphocytes (TILs). For those tumors that have been invaded by lymphocytes, they are profoundly exhausted and ineffective. While many immunotherapy approaches seek to reinvigorate immune cells at the tumor, this requires TILs to be present.
- immunotherapy
- glioblastoma
- T cells
- T lymphocytes
1. Introduction
Immune surveillance of the central nervous system (CNS) is essential for environmental homeostasis and pathogen clearance. Without immune surveillance, opportunistic infections in the CNS commonly develop [1]. However, the entry of immune cells into the CNS is tightly controlled by the blood–brain barrier (BBB) and the blood–cerebrospinal fluid (BCSF) barrier. These formidable barriers lack fenestrations, exhibit a low degree of pinocytosis, and are sealed together by a network of intracellular junctions [2,3]. While this close control is desirable in health to avoid runaway immune responses in the CNS, restricted immune cell entry severely hampers the effectiveness of immunotherapy in glioblastoma [4]. This is further complicated by the immunosuppressive tumor microenvironment (TME), which consists of endothelial cells, pericytes, fibroblasts, and regulatory immune cells [5]. The TME drives effector immune cell exhaustion, thereby shielding solid malignancies from immune attack [6]. While immune checkpoint inhibition (ICI) seeks to reverse this exhausted state and ‘release the brakes’ on regional T cells, it is notable that the evaluation of resected stage IV gliomas are either devoid or demonstrate limited numbers of tumor-infiltrating lymphocytes (TILs) [7,8]. This would suggest that ICI will struggle owing to the lack of targets to reinvigorate. Indeed, initial trials of ICI in glioblastoma have failed [9]. However, when ICI is combined with increased numbers of functional TILs in pre-clinical models, long-term survival can be achieved [10,11].
While the CNS does host several immune cell classes, including T cells, these immune cells are clustered away from the tumor-bearing parenchyma in regions such as the choroid plexus, the meninges (containing the subarachnoid and perivascular spaces), and the CSF [12,13,14,15,16,17]. The clinical implications of this clustering were recognized as long ago as 1923, where Murphy and Sturm confirmed Shirai’s initial finding that foreign tumors in the parenchyma could grow, but tumors implanted close to the ventricles (and thus the immune interfaces) were rejected [18]. Fortunately, immune responses in the CNS can be bolstered by an adaptive response originating from the periphery. Medawar demonstrated in 1948 that tumors implanted into brain tissue can be rejected following exposure to tumor antigens outside of the CNS [19]. Recruitment of peripheral T cells into the parenchyma also occurs in multiple sclerosis (MS) and its animal analogue experimental autoimmune encephalitis (EAE) [20].
Even though adaptive immune clearance of tumors is possible, glioblastoma possesses several mechanisms that suppress the recruitment and functioning of T cells. Glioblastoma expresses decreased levels of lymphangiogenesis-promoting factors such as VEGF-C, reducing potential routes for T cell ingress, while the highly immunosuppressive tumor microenvironment (TME) blunts the response of any lymphocytes that reach the tumor [21,22,23].
2. The Glia Limitans—Accessing the Parenchyma
3. T Cell Trafficking through the Parenchyma
Once past the glia limitans, effector T cells must reach and infiltrate the tumor to exert their cytotoxic effect. As discussed in the introduction, glioblastoma can restrict T cell trafficking due to the downregulated expression of VEGF-C, resulting in restricted lymphangiogenesis [22]. Notably, in patients treated with neoadjuvant anti-PD-1, VEGF-C expression was highly correlated with increased infiltration of T cells [138]. Thus, restoring levels of lymphangiogenesis-promoting factors such as VEGF-C could also enhance T cell homing and infiltration to the tumor. This is supported by the findings of Song et al., who demonstrated that intra-cisterna magna injections of an adeno-associated viral vector coding for VEGF-C could remodel meningeal lymphatic vessels in murine models of glioma [22]. Further enhanced expression of VEGF-C in lymphatic endothelial cells could potentiate the effect of checkpoint blockade due to enhanced T cell infiltration [22].
T cell motility is also dependent on metabolic pathways that are often usurped by rapidly proliferating tumors. Tumor cells demonstrate increased glucose uptake and lactose production, even in the presence of oxygen and functioning mitochondria (known as the Warburg effect) [139,140]. This affords the tumor and other rapidly proliferating cells essential anabolic precursors for cell proliferation [140]. The increased glucose demand by tumor cells therefore decreases the amount available for circulating T cells to maintain effector and migratory function [141]. Aerobic glycolysis is the main source of ATP production in leukocytes, which is required for the energetic demands of migration [142,143]. Inhibition of the T cell glycolytic pathway through administration of 2-DG and rapamycin causes a decrease in naïve T cell motility, demonstrating the importance of glucose in T cell homing [144,145]. The associated build-up of lactate caused by the Warburg effect also results in decreased migration of CD4 + T cells and a loss of cytolytic function of CD8 + T cells by interfering with T cell glycolysis [145,146,147,148]. However, this effect can be reversed, as demonstrated in an animal model of peritonitis where antibody-mediated blockade of lactate transporters on T cells allowed them to maintain their migratory potential [149]. Expression of CTLA-4 decreases the expression of the glucose transporter GLUT-1 on T cells, and further decreases effector function, implying that combinatorial approaches using checkpoint blockade may aid with T cell trafficking as well as reinvigoration of function [142,150]. However, recent work suggests that exhausted human CD8 + T cells may actually become more mobile [151]. CTLA-4 signaling can lead to a RAP1-mediated increase in LFA-1 binding, which can induce migration [152]. This has potential implications for considering which form of ICI would best work with a tumor where T cell trafficking poses a significant challenge. An overview of the metabolic pathways limiting T cell efficacy in glioblastoma is shown in Figure 2 .
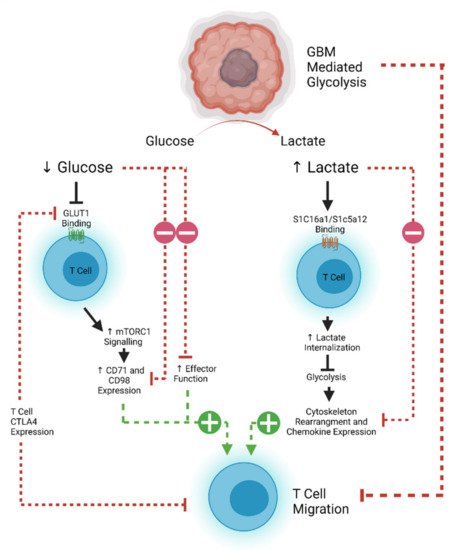
Another mediator of T cell glycolysis is the PI3K/AKT/mTOR pathway, whose activation can also downregulate the expression of adhesion and migration molecules CD62L, CCR7, and S1P1 in CD8 + T cells [142,153]. Loss of S1P1 has been shown to mediate T cell sequestration in bone marrow in glioblastoma, while S1P1 + cells are resistant to sequestration and can return into the circulation [142,154,155,156]. Therefore, reversing sequestration will be critical for future immunotherapy efficacy and is currently the subject of ongoing therapeutic investigation [157]. While one approach may be to inhibit the PI3K/AKT/mTOR pathway, this inhibition must be selective, as AKT possesses three isoforms which have varying pro- and anti-tumor effects. AKT signaling also plays an important role for the development of effector-like memory CD8 + T cells necessary for tumor immune surveillance [158]. Interestingly, recent work has described small-molecule inhibitors that may be capable of targeting pathogenic AKT isoforms only (AKT1 and AKT2) while leaving the tumor-suppressive functionality of AKT3 intact [159,160]. Indeed, specific AKT1 and 2 inhibition has been associated with enhanced central memory CD8 + T cell proliferation with prolonged cytokine and Granzyme B production, making this a potential future therapeutic strategy [158,159,160,161].
4. The Tumor Microenvironment
Once T cells traffic past the BBB and through the parenchyma, they will encounter the highly immunosuppressive tumor microenvironment. This is made up of regulatory T cells (CD4 + CD25 + FOXP3 + ), tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs), as well as other stromal cells such as GSC-derived pericytes [23,162]. These can all work to suppress effector T cell function. Regulatory T cells induce T cell exhaustion and apoptosis, signaling via programmed death-ligand 1 (PD-L1), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), lymphocyte activation gene 3 (LAG-3) , T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), and others [163,164]. They also can dampen the production of inflammatory cytokines and CTL proliferation by downregulating interleukin-2 and interferon-γ [165]. Gliomas are adept at recruiting regulatory T cells to the microenvironment by over-production of factors such as indoleamine 2,3 -dioxygenase-1 (IDO-1) [166]. As mentioned previously, GSC-derived pericytes also secrete CCL5, which can promote the recruitment of regulatory T cells to the TME [54]. Stromal cells in the microenvironment also produce highly immunosuppressive cytokines, such as transforming growth factor β (TGFβ) and interleukin-10 (IL-10) [167,168].
Despite the numerous targets for blockade, it is notable that ICI and the interruption of pro-tumor metabolic pathways have failed as a monotherapy [9,169]. Increasingly, attention is turning towards combinatorial therapies, where multiple drivers of T cell exhaustion can be blocked simultaneously [170]. This includes using bispecific antibodies against TGF-β and PD-L1 or against PD-L1 and the anti-agonist CD27 [171,172,173]. These approaches are currently being evaluated in Phase I trials in advanced solid tumors (NCT04429542, NCT04440943). Cytokine modulation approaches are also a potential avenue for enhancing T cell activity in the TME, as seen in ‘armored’ CAR-T constructs. The addition of IL-12, IL-15, or IL-18 along with antigen specificity to T cells appears to result in greater CTL activity and anti-tumor efficacy [174,175,176]. A high percentage of regulatory T cells in the peripheral blood of GBM patients express CCR4 compared to controls (74 vs. 43%) [135]. CCL4 binds CCL22 (and others), which has been shown to be overexpressed in freshly resected human glioma cells, and blockade of CCR4 in vitro can significantly reduce regulatory T cell migration [135]. Targeting fibroblast activation proteins or introducing heparinase-expressing agents may also help to disrupt immunosuppressive stromal elements [146,177,178]. Intratumoral APCs are also necessary to stimulate and retain infiltrating lymphocytes at the tumor site, as well as carrying antigens to draining lymph nodes and cross priming peripheral CD8 T cells [179,180,181]. The administration of intratumoral FMS-like tyrosine kinase 3 ligand (Flt3L) and poly I:C has been shown to expand and mature dendritic cell precursors, resulting in greater antitumor efficacy when combined with immunotherapies such as PD-L1 blockade or oncolytic herpes simplex viruses [179,182].
Standard-of-care therapies also can help drive a more potent immune response. Temozolomide (TMZ) is an alkylating chemotherapy whose main function is to induce DNA double-stranded breaks, resulting in tumor cell death [183]. Interestingly, TMZ can also help to reduce the numbers of peripheral regulatory T cells, as well as interrupting their migration [136,184]. In disease states such as glioblastoma, tumor cells and platelet-derived growth factor receptor beta (PDGFRβ)-expressing cells of the neurovascular sub-units (such as pericytes and perivascular fibroblast-like cells) produce CCL2 to recruit regulatory T cells and dampen the effector response [185]. TMZ interrupts the CCL2–CCR4 axis, thereby reducing this effect [136,184]. Combining immunotherapy with radiotherapy also can help to polarize the T cell response to a cytotoxic phenotype by inducing greater T cell receptor diversity and expanding the numbers of tumor-infiltrating lymphocytes and effector memory T cells [186]. In pre-clinical murine models of glioma, radiotherapy combined with antibodies against markers of exhaustion such as TIM-3 and PD-1 was able to produce long-term survival [11].
This entry is adapted from the peer-reviewed paper 10.3390/cancers13215367