Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Pathology
Nitric oxide (NO), carbon monoxide (CO), and hydrogen sulfide (H2S), three major gasotransmitters, are involved in pleiotropic biofunctions. Research on their roles in hypertension and kidney disease has greatly expanded recently. The developing kidney can be programmed by various adverse in utero conditions by so-called renal programming, giving rise to hypertension and kidney disease in adulthood.
- kidney disease
- gasotransmitter
- carbon monoxide
- hypertension
- developmental origins of health and disease (DOHaD)
- hydrogen sulfide
- asymmetric dimethylarginine
- heme oxygenase
- nitric oxide
1. Introduction
Gasotransmitters, such as nitric oxide (NO), carbon monoxide (CO), and hydrogen sulfide (H2S), are small gaseous molecules that penetrate membranes and play key roles in biology. Although these gases are toxic in excess, they are endogenously generated and exert specific biological functions at the physiological level [1][2][3]. A brief overview of their toxic and physiological levels is given in Table 1 [1][2][3]. Since it was identified as the endothelium-derived relaxing factor in the 1980s, NO has rapidly gained attention as one of the most important signaling molecules in the cardiovascular system [4]. A decade later, CO emerged as a gaseous vascular modulator of the cardiovascular system [5]. H2S, next to NO and CO, has emerged as a third gasotransmitter with key roles in the regulation of cardiovascular and other systems [6]. All three gases have a significant impact on human health and potential value as a therapeutic target [1][2][4].
Table 1. Physiological and toxic levels of NO, CO, and H2S.
|
Physiological Level |
Toxic Level |
||
|---|---|---|---|
|
Gas |
Blood Concentration |
Exposure Limit |
IDLH |
|
Nitric oxide (NO) |
Low nM |
TMA 25 ppm |
100 ppm |
|
Carbon monoxide (CO) |
nM–μM |
TMA 35 ppm/C 200 ppm |
100 ppm |
|
Hydrogen sulfide (H2S) |
High nM–low μM |
C 10 ppm |
1200 ppm |
TWA = Time-weighted average; C = Ceiling; IDLH = Immediately dangerous to life or health concentrations; ppm = parts per million.
Chronic kidney disease (CKD) and hypertension are major non-communicable diseases, which are the leading causes of global deaths. According to the WHO, one in five women and one in four men have hypertension worldwide [7]. An estimated ~10% of the global population has CKD [8]. Hypertension and CKD are closely associated with an overlapping and interlinked cause and effect relationship [9], such that hypertension can lead to CKD progression and CKD is the most common cause of secondary hypertension. Of note, hypertension as well as kidney disease can take their origins in early life, and when identified early, can be healed to prevent more associated disorders and serious complications.
During kidney development, various early-life adverse environmental conditions can lead to hypertension and kidney disease in adulthood [10]. The idea was recently named “Developmental Origins of Health and Disease” (DOHaD) [11]. Conversely, through shifting therapeutic approach from adulthood to early life, namely, reprogramming, we have the potential to revoke disease processes before disease becomes apparent [12][13].
2. Implications of Gasotransmitters in Pregnancy
A variety of adverse conditions during pregnancy can affect fetal development resulting in hypertension and kidney disease in adult offspring, including maternal malnutrition, maternal exposure to environmental chemicals/toxins, maternal illnesses, medication uses in pregnancy, etc. [10][12][13][14][15][16]. Gasotransmitters play a crucial role in the regulation of maternal hemodynamics, placenta vascular development, embryogenesis, feto-placental vascular reactivity, and fetal development during pregnancy [17][18][19]. Abnormalities of gasotransmitter production and signaling in compromised pregnancy are linked to adverse pregnancy and fetal outcomes. A drawing schematic summarizing the enzymatic production of NO, CO, and H2S, and signaling pathways able to maintain normal pregnancy and fetal development are depicted in Figure 1. Each gasotransmitter is discussed in turn.
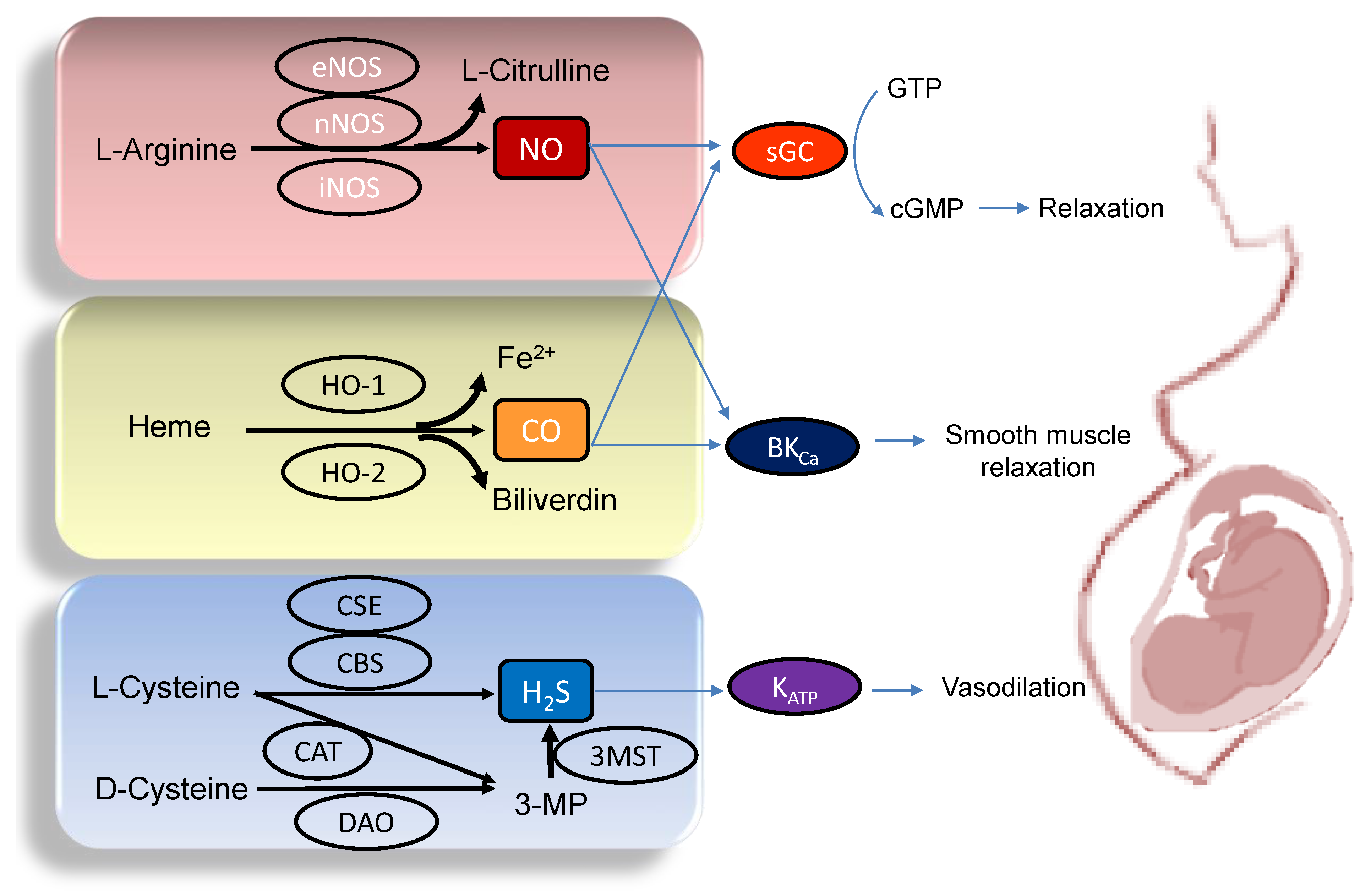
Figure 1. Schematic illustration of the enzymatic synthesis of NO, CO, and H2S and downstream signaling able to maintain maternal and feto-placental homeostasis. NO (upper panel) is formed by neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS) from L-Arginine. Heme oxygenase-1 (HO-1) and -2 (HO-2) enzymes degrade heme to generate CO, iron, and biliverdin (middle panel). Three enzymes have been identified to enzymatically generate H2S (Lower panel), cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), and 3-mercaptopyruvate sulphurtransferase (3MST). CBS and CSE produce H2S using L-cysteine. In an alternative pathway, 3-mercaptopyruvate (3-MP), the substrate for 3MST to produce H2S, is provided by cysteine aminotransferase (CAT) using L-Cysteine and D-amino acid oxidase (DAO) using D-Cysteine, respectively. The blue arrow lines indicate downstream signals of gasotransmitters in the maintenance of homeostasis in pregnancy. NO and CO both can activate soluble guanylate cyclase (sGC) to increase cGMP, resulting in smooth muscle relaxation. The large-conductance Ca2+-activated K+ channel (BKCa) can also be regulated by NO and CO to elicit vasodilatation. Additionally, through the activation of ATP-sensitive K+-channels (KATP), H2S can cause vasodilation in pregnancy.
3. Implications of Gasotransmitters in Hypertension and Kidney Disease
3.1. Gasotransmitters and Hypertension
Several lines of evidence indicate that NO, CO, and H2S play key roles in the pathogenesis of hypertension. The first are observations on knockout mice lacking genes responsible for gasotransmitter synthesis. First, eNOS knockout mice displayed hypertension [20]. The importance of H2S-generating enzymes in hypertension has also been demonstrated using CSE, CBS, or 3MST knockout mice [21][22][23][24]. Another report showed male HO-2 knockout mice are prone to develop renovascular hypertension [25].
The second line of evidences report dysregulated gasotransmitter signaling pathways in human and experimental models of hypertension. Prior research has addressed impaired L-Arginine–ADMA–NO pathway in the development of hypertension [26]. Dysregulated HO-1–CO pathway was reported to induce vascular dysfunction and hypertension in various animal models [27]. Likewise, deficiencies in H2S-generating enzymes and/or activity in hypertension has been established in various animal models, including the NO-deficient rats [28], the Dahl salt-sensitive rats [29], the spontaneously hypertensive rat (SHR) [30], and the renovascular hypertensive model [31].
3.2. Gasotransmitters and Kidney Disease
The gasotransmitter generating enzymes iNOS, eNOS, nNOS, HO-1, HO-2, CSE, CBS, and 3MST were detected in kidney cells comprising podocytes, glomerular endothelial cells, tubular cells, and mesangial cells, but not all of them are constitutively expressed in every cell type [35][36]. For example, eNOS is expressed in the glomerular endothelial cells, peritubular capillaries, and vascular bundles, while nNOS is mainly detected in the tubular epithelial cells of the macula densa [36]. Of note, iNOS and HO-1 are not constitutively expressed in the kidney but only expressed under certain pathophysiological conditions like inflammation [36].
In the kidney, NO performs important signaling functions including the modulation of renal sympathetic neural activity, control of renal hemodynamics, regulation of pressure-natriuresis, blunting of tubuloglomerular feedback, and inhibition of tubular sodium reabsorption [36]. Accordingly, impaired NO signaling has been implicated in the pathogenesis of kidney diseases. As reviewed elsewhere [37][38], kidney injury is attributed to NO deficiency in a variety of CKD models, such as diabetic nephropathy, chronic glomerular nephritis, the 5/6 nephrectomy model, the aging kidney, the Zucker obese rat, chronic allograft nephropathy, etc.
The beneficial actions of CO in the kidney have also been recognized [39]. Inhibition of superoxide production, activation of sGC, stimulation of NO production, and stimulation of p38 mitogen-activated protein kinase (MAPK) pathway are all examples of the beneficial effects of CO in the kidney to protect the kidney [39]. Deficiency or inhibition of HO-1 in animal models worsens renal structure and function, while increased expression is protective [40]. So far, evidences from animal models indicate that several kidney diseases have been associated with impaired HO-1 or -2 system, including diabetic nephropathy [41], lupus nephritis [42], nephrotoxic nephritis [43], ischemia-reperfusion injury [44], obstructive nephropathy [45], and CKD [46].
H2S regulates basic physiologic mechanisms of the kidney such as sodium reabsorption, glomerular filtration, and renal homeostasis [47]. In some animal models of kidney disease, such as CKD [48], acute kidney injury [49], cisplatin nephropathy [50], obstructive nephropathy [51], and diabetic nephropathy [52], it can serve as an agent that ameliorates kidney injury.
3.3. Crosstalk between NO, CO, and H2S in the Kidney and BP Control
Although NO and H2S share the same sGC–cGMP pathway to elicit relaxation in kidney cells [53], they act at different levels, with NO increasing production of cGMP through stimulation of sGC and H2S inhibiting cGMP degradation [54]. In rats, inhibition of NO by L-NAME, causes hypertension that can be prevented by the administration of sodium hydrosulfide (NaHS, a H2S donor), which also rescues NO bioavailability [55]. These data support the notion that there exists a NO/H2S crosstalk in the control of blood pressure (BP).
One of the NO-based cellular signaling pathways is via protein S-nitrosylation, the covalent addition of NO moiety to the sulfur atom of cysteine residues [56]. S-nitrosylation of specific proteins has been shown to be protective against kidney injury [56]. As observed for NO, H2S also employs post-translational modifications, namely, S-sulfhydration [57]. Endogenous H2S physiologically S-sulfydrates proteins on the thiol group of cysteine residues (e.g., glutathione), leading to the formation of the –SSH moiety. These observations lead to a hypothesis that there might be competition between S-nitrosylation and S-sulfhydration for the same cysteine residues in proteins, thus allowing the two gasotransmitters to regulate each other [54].
CO could also target sGC and regulate NO-mediated vasodilatation [58], which was supported by a report showing that transgenic mice overexpressing cell-specific HO-1 exhibit hypertension coinciding with decreased cGMP production in response to NO [59]. Additionally, CO could interfere with NOS activity and reduce NO generation as a consequence, thereby limiting NO-mediated vasodilation [60]. Although much is known about the CO and NO signaling pathways in the kidney, we so far do not fully understand how these two gaseous signaling systems interact with each other.
Moreover, all three gasotransmitters are involved in activation of nuclear factor erythroid 2-related factor 2 (NRF2) (Figure 2). NRF2 is a major regulator of HO-1 transcription responding to oxidative stress [40]. Upon activation, the NRF2-HO-1 pathway protects chronic kidney disease progression related to reduction of oxidative stress, inhibition of transforming growth factor-β (TGF-β)-driven fibrosis, reduction of inflammation and apoptosis [61]. Under basal conditions, NRF2 levels are kept low through the interaction with Kelch-like ECH associated protein 1 (KEAP1). Upon binding, the NRF2-KEAP1 interaction stabilizes the complex allowing for ubiquitylation, and ultimately proteasomal degradation of NRF2 [61]. Of note, NO and H2S can activate NRF2 via S-nitrosylation and S-sulfhydration of KEAP1, respectively [62][63]. In addition to NRF2, NO can regulate other redox-regulatory transcription factors, like nuclear factor κB (NFκB) and hypoxia-inducible factor-1α (HIF-1α), via S-nitrosylation [62]. As NFκB mediates inflammation and HIF-1α induces HO-1 expression, NO can interact with the NRF2–HO-1–CO signaling pathway in many different ways to prevent CKD progression. NFκB can also be S-sulfhydrated by H2S [63]. These observations indicate crosstalk mechanisms between NO, CO, and H2S are important determinants for kidney disease (Figure 2).
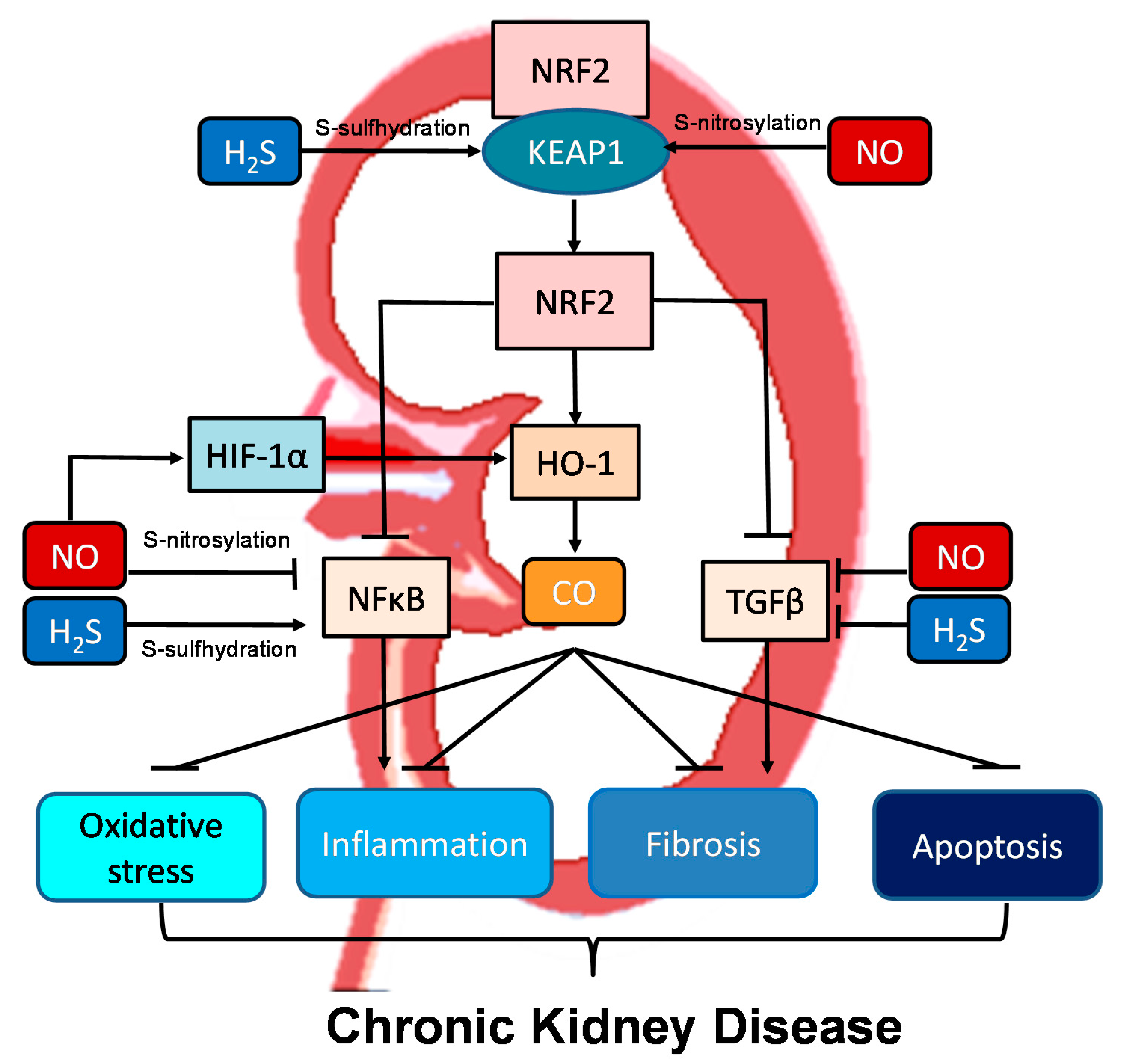
Figure 2. Schematic illustration of the crosstalk among NO, CO, and H2S and downstream nuclear factor erythroid 2-related factor 2 (NRF2)–heme oxygenase-1 (HO-1) signaling able to protect chronic kidney disease progression. Activation of the NRF2–HO-1–CO signaling pathway suppresses nuclear factor κB (NFκB) and transforming growth factor-β (TGF-β), consequently inhibiting oxidative stress, inflammation, fibrosis, and apoptosis. NO and H2S can activate NRF2 via S-nitrosylation and S-sulfhydration of Kelch-like ECH associated protein 1 (KEAP1), respectively. Via S-nitrosylation, NO can also inhibit NFκB-induced inflammation and activate hypoxia-inducible factor-1α (HIF-1α), a HO-1 inducer. Additionally, NFκB can also be regulated by H2S via S-sulfhydration. NO and H2S also reduce TGFβ-mediated fibrosis.
Although the beneficial actions of gasotransmitters against established kidney disease and hypertension have been established, their roles in mediating programmed responses behind developmental origins remain unclear. For this reason, this review will next outline the potential early-life interventions targeting NO, CO, and H2S signaling that may pose new opportunities for the therapeutic protection of hypertension and kidney disease.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22157808
References
- Kajimura, M.; Fukuda, R.; Bateman, R.M.; Yamamoto, T.; Suematsu, M. Interactions of multiple gas-transducing systems: Hallmarks and uncertainties of CO, NO, and H2S gas biology. Antioxid. Redox Signal. 2010, 13, 157–192.
- Polhemus, D.J.; Lefer, D.J. Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ. Res. 2014, 114, 730–737.
- NIOSH Pocket Guide to Chemical Hazards. 2020. Available online: https://www.cdc.gov/niosh/npg/ (accessed on 19 July 2021).
- Lancaster, J.R. Historical origins of the discovery of mammalian nitric oxide (nitrogen monoxide) production/physiology/pathophysiology. Biochem. Pharmacol. 2020, 176, 113793.
- Durante, W. Carbon monoxide and bile pigments: Surprising mediators of vascular function. Vasc. Med. 2002, 7, 195–202.
- Kimura, H. The physiological role of hydrogen sulfide and beyond. Nitric Oxide 2014, 41, 4–10.
- World Health Organization. Hypertension. 2019. Available online: https://www.who.int/news-room/fact-sheets/detail/hypertension (accessed on 12 May 2021).
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128.
- Weir, M.R. Hypertension and the kidney: Perspectives on the relationship of kidney disease and cardiovascular disease. Clin. J. Am. Soc. Nephrol. 2009, 4, 2045–2050.
- Tain, Y.L.; Hsu, C.N. Developmental origins of chronic kidney disease: Should we focus on early life? Int. J. Mol. Sci. 2017, 18, 381.
- Haugen, A.C.; Schug, T.T.; Collman, G.; Heindel, J.J. Evolution of DOHaD: The impact of environmental health sciences. J. Dev. Orig. Health Dis. 2015, 6, 55–64.
- Tain, Y.L.; Joles, J.A. Reprogramming: A preventive strategy in hypertension focusing on the kidney. Int. J. Mol. Sci. 2016, 17, 23.
- Paauw, N.D.; van Rijn, B.B.; Lely, A.T.; Joles, J.A. Pregnancy as a critical window for blood pressure regulation in mother and child: Programming and reprogramming. Acta Physiol. 2017, 219, 241–259.
- Chong, E.; Yosypiv, I.V. Developmental programming of hypertension and kidney disease. Int. J. Nephrol. 2012, 2012, 760580.
- Paixão, A.D.; Alexander, B.T. How the kidney is impacted by the perinatal maternal environment to develop hypertension. Biol. Reprod. 2013, 89, 144.
- Hsu, C.N.; Tain, Y.L. Animal models for DOHaD research: Focus on hypertension of developmental origins. Biomedicines 2021, 9, 623.
- Holwerda, K.M.; Faas, M.M.; van Goor, H.; Lely, A.T. Gasotransmitters: A solution for the therapeutic dilemma in preeclampsia? Hypertension 2013, 62, 653–659.
- Guerra, D.D.; Hurt, K.J. Gasotransmitters in pregnancy: From conception to uterine involution. Biol. Reprod. 2019, 101, 4–25.
- Rengarajan, A.; Mauro, A.K.; Boeldt, D.S. Maternal disease and gasotransmitters. Nitric Oxide 2020, 96, 1–12.
- Huang, P.L.; Huang, Z.; Mashimo, H.; Bloch, K.D.; Moskowitz, M.A.; Bevan, J.A.; Fishman, M.C. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 1995, 377, 239–242.
- Shibuya, N.; Kimura, H. Production of hydrogen sulfide from d-cysteine and its therapeutic potential. Front. Endocrinol. 2013, 4, 87.
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science 2008, 322, 587–590.
- Dayal, S.; Bottiglieri, T.; Arning, E.; Maeda, N.; Malinow, M.R.; Sigmund, C.D.; Heistad, D.D.; Faraci, F.M.; Lentz, S.R. Endothelial dysfunction and elevation of S-adenosylhomocysteine in cystathionine beta-synthase-deficient mice. Circ. Res. 2001, 88, 1203–1209.
- Mani, S.; Li, H.; Untereiner, A.; Wu, L.; Yang, G.; Austin, R.C.; Dickhout, J.G.; Lhoták, Š.; Meng, Q.H.; Wang, R. Decreased Endogenous Production of Hydrogen Sulfide Accelerates Atherosclerosis. Circulation 2013, 127, 2523–2534.
- Stout, J.M.; Gousset, M.U.; Drummond, H.A.; Gray, W., 3rd; Pruett, B.E.; Stec, D.E. Sex-specific effects of heme oxygenase-2 deficiency on renovascular hypertension. J. Am. Soc. Hypertens. 2013, 7, 328–335.
- Tain, Y.L.; Hsu, C.N. Targeting on asymmetric dimethylarginine-related nitric oxide-reactive oxygen species imbalance to reprogram the development of hypertension. Int. J. Mol. Sci. 2016, 17, 2020.
- Durante, W. Targeting heme oxygenase-1 in vascular disease. Curr. Drug Targets 2010, 11, 1504–1516.
- Jin, S.; Teng, X.; Xiao, L.; Xue, H.; Guo, Q.; Duan, X.; Chen, Y.; Wu, Y. Hydrogen sulfide ameliorated L-NAME-induced hypertensive heart disease by the Akt/eNOS/NO pathway. Exp. Biol. Med. (Maywood). 2017, 242, 1831–1841.
- Huang, P.; Chen, S.; Wang, Y.; Liu, J.; Yao, Q.; Huang, Y.; Li, H.; Zhu, M.; Wang, S.; Li, L.; et al. Down-regulated CBS/H2S pathway is involved in high-salt-induced hypertension in Dahl rats. Nitric Oxide 2015, 46, 192–203.
- Xiao, L.; Dong, J.-H.; Jing-Hui, D.; Xue, H.-M.; Guo, Q.; Teng, X.; Wu, Y.-M. Hydrogen sulfide improves endothelial dysfunction via downregulating BMP4/COX-2 pathway in rats with hypertension. Oxid. Med. Cell. Longev. 2016, 2016, 1–10.
- Van Goor, H.; Born, J.C.V.D.; Hillebrands, J.-L.; Joles, J.A. Hydrogen sulfide in hypertension. Curr. Opin. Nephrol. Hypertens. 2016, 25, 107–113.
- Paulo, M.; Costa, D.E.F.R.; Bonaventura, D.; Lunardi, C.N.; Bendhack, L.M. Nitric oxide donors as potential drugs for the treatment of vascular diseases due to endothelium dysfunction. Curr. Pharm. Des. 2020, 26, 3748–3759.
- Ndisang, J.F.; Tabien, H.E.; Wang, R. Carbon monoxide and hypertension. J. Hypertens. 2004, 22, 1057–1074.
- Wen, Y.-D.; Wang, H.; Zhu, Y.Z. The Drug developments of hydrogen sulfide on cardiovascular disease. Oxid. Med. Cell. Longev. 2018, 2018, 4010395.
- Beck, K.F.; Pfeilschifter, J. Gasotransmitter synthesis and signalling in the renal glomerulus. Implications for glomerular diseases. Cell. Signal. 2021, 77, 109823.
- Kone, B.C. Nitric oxide synthesis in the kidney: Isoforms, biosynthesis, and functions in health. Semin. Nephrol. 2004, 24, 299–315.
- Baylis, C. Nitric oxide synthase derangements and hypertension in kidney disease. Curr. Opin. Nephrol. Hypertens. 2012, 21, 1–6.
- Wilcox, C.S. Oxidative stress and nitric oxide deficiency in the kidney: A critical link to hypertension? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R913–R935.
- Csongradi, E.; Juncos, L.A.; Drummond, H.A.; Vera, T.; Stec, D.E. Role of carbon monoxide in kidney function: Is a little carbon monoxide good for the kidney? Curr. Pharm. Biotechnol. 2012, 13, 819–826.
- Lever, J.M.; Boddu, R.; George, J.F.; Agarwal, A. Heme Oxygenase-1 in Kidney Health and Disease. Antioxid. Redox Signal. 2016, 25, 165–183.
- Ndisang, J.F.; Jadhav, A. Hemin therapy improves kidney function in male streptozotocin-induced diabetic rats: Role of the heme oxygenase/atrial natriuretic peptide/adiponectin axis. Endocrinology 2014, 155, 215–229.
- Mackern-Oberti, J.P.; Llanos, C.; Carreno, L.J.; Riquelme, S.A.; Jacobelli, S.H.; Anegon, I.; Kalergis, A.M. Carbon monoxide exposure improves immune function in lupus-prone mice. Immunology 2013, 140, 123–132.
- Mosley, K.; Wembridge, D.E.; Cattell, V.; Cook, H.T. Heme oxygenase is induced in nephrotoxic nephritis and hemin, a stimulator of heme oxygenase synthesis, ameliorates disease. Kidney Int. 1998, 53, 672–678.
- Ferenbach, D.A.; Ramdas, V.; Spencer, N.; Marson, L.; Anegon, I.; Hughes, J.; Kluth, D.C. Macrophages expressing heme oxygenase-1 improve renal function in ischemia/reperfusion injury. Mol. Ther. 2010, 18, 1706–1713.
- Kie, J.H.; Kapturczak, M.H.; Traylor, A.; Agarwal, A.; Hill-Kapturczak, N. Heme oxygenase-1 deficiency promotes epithelial-mesenchymal transition and renal fibrosis. J. Am. Soc. Nephrol. 2008, 19, 1681–1691.
- Zager, R.A.; Johnson, A.C.; Becker, K. Acute unilateral ischemic renal injury induces progressive renal inflammation, lipid accumulation, histone modification, and ‘‘end-stage’’ kidney disease. Am. J. Physiol. Renal Physiol. 2011, 301, F1334–F1345.
- Scammahorn, J.J.; Nguyen, I.T.N.; Bos, E.M.; Van Goor, H.; Joles, J.A. Fighting oxidative stress with sulfur: Hydrogen sulfide in the renal and cardiovascular systems. Antioxidants 2021, 10, 373.
- Dugbartey, G.J. The smell of renal protection against chronic kidney disease: Hydrogen sulfide offers a potential stinky remedy. Pharm. Rep. 2018, 70, 196–205.
- Chen, Y.; Jin, S.; Teng, X.; Hu, Z.; Zhang, Z.; Qiu, X.; Tian, D.; Wu, Y. Hydrogen sulfide attenuates LPS-induced acute kidney injury by inhibiting inflammation and oxidative stress. Oxid. Med. Cell. Longev. 2018, 2018, 6717212.
- Cao, X.; Zhang, W.; Moore, P.K.; Bian, J. Protective smell of hydrogen sulfide and polysulfide in cisplatin-induced nephrotoxicity. Int. J. Mol. Sci. 2019, 20, 313.
- Lin, S.; Visram, F.; Liu, W.; Haig, A.; Jiang, J.; Mok, A.; Lian, D.; Wood, M.E.; Torregrossa, R.; Whiteman, M.; et al. GYY4137, a slow-releasing hydrogen sulfide donor, ameliorates renal damage associated with chronic obstructive uropathy. J. Urol. 2016, 196, 1778–1787.
- Xue, R.; Hao, D.D.; Sun, J.P.; Li, W.W.; Zhao, M.M.; Li, X.H.; Chen, Y.; Zhu, J.H.; Ding, Y.J.; Liu, J.; et al. Hydrogen sulfide treatment promotes glucose uptake by increasing insulin receptor sensitivity and ameliorates kidney lesions in type 2 diabetes. Antioxid. Redox Signal. 2013, 19, 5–23.
- Feliers, D.; Lee, H.J.; Kasinath, B.S. Hydrogen sulfide in renal physiology and disease. Antioxid. Redox Signal. 2016, 25, 720–731.
- Cirino, G.; Vellecco, V.; Bucci, M. Nitric oxide and hydrogen sulfide: The gasotransmitter paradigm of the vascular system. Br. J. Pharmacol. 2017, 174, 4021–4031.
- Zhong, G.; Chen, F.; Cheng, Y.; Tang, C.; Du, J. The role of hydrogen sulfide generation in the pathogenesis of hypertension in rats induced by inhibition of nitric oxide synthase. J. Hypertens. 2003, 21, 1879–1885.
- Zhou, H.L.; Zhang, R.; Anand, P.; Stomberski, C.T.; Qian, Z.; Hausladen, A.; Wang, L.; Rhee, E.P.; Parikh, S.M.; Karumanchi, S.A.; et al. Metabolic reprogramming by the S-nitroso-CoA reductase system protects against kidney injury. Nature 2019, 565, 96–100.
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K.; Barrow, R.K.; Yang, G.; Wang, R.; Snyder, S.H. H2S signals through protein S-sulfhydration. Sci. Signal. 2009, 2, ra72.
- Stec, D.E.; Drummond, H.A.; Vera, T. Role of carbon monoxide in blood pressure regulation. Hypertension 2008, 51, 597–604.
- Imai, T.; Morita, T.; Shindo, T.; Nagai, R.; Yazaki, Y.; Kurihara, H.; Suematsu, M.; Katayama, S. Vascular smooth muscle cell-directed overexpression of heme oxygenase-1 elevates blood pressure through attenuation of nitric oxide-induced vasodilation in mice. Circ. Res. 2001, 89, 55–62.
- Thorup, C.; Jones, C.L.; Gross, S.S.; Moore, L.C.; Goligorsky, M.S. Carbon monoxide induces vasodilation and nitric oxide release but suppresses endothelial NOS. Am. J. Physiol. 1999, 277, F882–F889.
- Uddin, M.J.; Kim, E.H.; Hannan, M.A.; Ha, H. Pharmacotherapy against oxidative stress in chronic kidney disease: Promising small molecule natural products targeting Nrf2-HO-1 signaling. Antioxidants 2021, 10, 258.
- Fernando, V.; Zheng, X.; Walia, Y.; Sharma, V.; Letson, J.; Furuta, S. S-Nitrosylation: An emerging paradigm of redox signaling. Antioxidants 2019, 8, 404.
- Iciek, M.; Kowalczyk-Pachel, D.; Bilska-Wilkosz, A.; Kwiecień, I.; Górny, M.; Włodek, L. S-sulfhydration as a cellular redox regulation. Biosci. Rep. 2015, 36, e00304.
This entry is offline, you can click here to edit this entry!