Arrhythmogenic cardiomyopathy (ACM) is a rare inherited cardiomyopathy characterized as fibro-fatty replacement, and a common cause for sudden cardiac death in young athletes. Development of heart failure (HF) has been an under-recognized complication of ACM for a long time. The current clinical management guidelines for HF in ACM progression have nowadays been updated. The current clinical management guidelines for HF in ACM progression have nowadays been updated. A comprehensive review for this great achievement in our understanding of HF in ACM is necessary.
- arrhythmogenic ventricular cardiomyopathy
- heart failure
- risk stratification
- prognosis
1. Introduction
Arrhythmogenic cardiomyopathy (ACM) is a familial heart disease with a prevalence of approximately 1:5000 [1][2][3]. The disease is a major cause of sudden cardiac death (SCD) in adolescents and young adults, especially during athletic activity. With the establishment of clinical risk prediction models and the use of implantable cardioverter-defibrillators (ICD) in high-risk patients, the incidence of malignant arrhythmic events has gradually reduced [4][5][6][7][8]. ACM is known to be a progressive disease, and with advanced right ventricular (RV) involvement and/or left ventricular (LV) involvement, symptoms and signs of heart failure (HF) may occur during later stages of disease. Despite the fact that the focus in ACM was on arrhythmias in earlier studies, various HF phenotypes have received attention more recently.
The prevalence and severity of progressive HF in ACM have been somewhat controversial, as the epidemiological and clinical characteristics of the disease varied greatly among reports from different centers [9][10][11][12]. In many retrospective clinical studies, HF was reported to be rare and often related to later stages of disease [13][14]. On the other hand, HF was also reported to be an early, and even first, manifestation of disease in other studies. In particular, it has even been recognized as one of the main causes for cardiac death and heart transplantation (HTx) [10][12].
The HF course of ACM is unique from other cardiac diseases, such as dilated cardiomyopathy and hypertrophic cardiomyopathy, which are more common causes for severe HF related adverse outcomes. The sequence and origin of HF symptoms in ACM is quite distinctive. The symptoms of HF often appear after the stage of electrophysiological disorder and originate from the right side pulmonary circulatory system in ACM. The severity of ACM has gender specificity and age differences, which could lead to different risk stratification from other cardiomyopathies. Apart from that, the genotype and exercise could also affect the HF progression in ACM, which requires a more specific prevention and treatment strategy in its clinical management.
In this entry, in order to provide a more comprehensive understanding of clinical phenotypes and management strategies, we report the prevalence, clinical course, risk stratification, prevention and treatment strategies of HF in ACM patients. Moreover, we propose our view of prospective research directions in this field.
2. Prevalence of HF in ACM
Progressive disease and occurrence of HF was described in various ACM registries across different ethnic backgrounds. HF has been reported from all ACM cohorts across the globe ( Figure 1 ). However, the prevalence of HF in this disease was reported differently in different centers because of the lack of consensus on the definition of HF and due to different inclusion methods. The clinical diagnosis of HF was mainly based on a combination of the American College of Cardiology/American Heart Association HF staging system and physician experience in different registries. The incidence of HF was reported to be in the range of 5% (defined by volume overload) to 49% (defined by the clinical symptoms and the severity of ventricular remodeling in echocardiogram or cardiac MRI imagines) among different centers and studies [10][15]. In the clinical reports from studies which only include the ACM probands, the incidence of HF was higher compared to some others which also enrolled their at-risk relatives. The incidence of HF in ACM was reported to be around 0.5% annually in primary care hospitals, which was much lower than that reported by tertiary care centers [16]. The causes of this difference may mainly derive from the unavoidable selection bias of the ACM population, in that patients with advanced HF were more likely treated in tertiary hospitals.
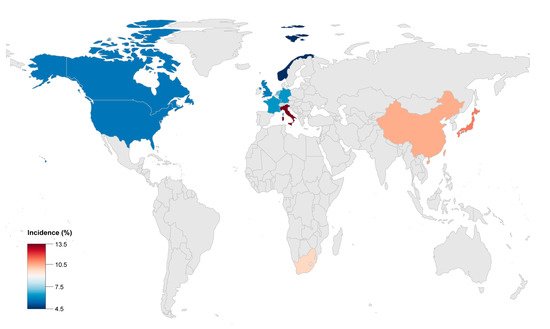
The global incidence of adverse outcomes, including HTx and cardiac death caused by HF in ACM, was reported to be 2 to 22% ( Table 1 ). Advanced HF in ACM was more common in Asia and could cause a poorer prognosis compared with Europe or America. The ACM patients from China or Japan had higher risk for HF related rehospitalization, heart transplantation and death. This could possibly be caused by the different proportion of certain gene mutations, such as plakophilin 2 ( PKP2 ) and Desmoglein 2 ( DSG2 ).
| Author | Publication Year | Study Population | Enrollment Period | Median Follow-Up (Years) | Male (%) | Endpoint | Incidence (%) | Malignant Arrythmias Events |
|---|---|---|---|---|---|---|---|---|
| Stefan Peters [17] | 1999 | 121 | 1986–1998 | 12.0 | 52.9 | 1 HTx/3 death | 3.3 | 14 RBBB |
| Jean-Sébastien Hulot [18] | 2004 | 130 | 1977–2000 | 8.1 | 76.9 | 14 death | 10.8 | 7 SCD, 17 SCA, 8 VF, 11 supraventricular arrhythmias |
| K Lemola [19] | 2005 | 61 | 4.6 | 72.1 | 5 HTx/2 death | 11.5 | 8 SCD, 8 SCA, 46 VT, 44 VT+LBBB | |
| Darshan Dalal [20] | 2005 | 100 | 6.0 | 51.0 | 2 HTx/1 death | 3 | 22 SCD, 1 SCA, 51 VT+LBBB | |
| Stefan Peters [15] | 2007 | 313 | 1986–2004 | 8.5 | 62.9 | 2 HTx/5 death | 2.2 | 5 SCD, 21 SCA, 96 SVT, 38 atrial arrhythmias |
| David A. Watkins [21] | 2009 | 50 | 2004–2009 | 4.6 | 66.0 | 2 HTx/3 death | 10 | 2 SCA, 41VT+LBBB |
| Bruno Pinamonti [22] | 2011 | 96 | 1976–2008 | 10.7 | 70.8 | 7 HTx/6 death | 13.5 | 3 SCA, 4 RBBB, 19 VT+LBBB, 4 atrial arrhythmias, 19 supraventricular arrhythmias |
| Masatoshi Komura [12] | 2010 | 35 | 4.5 | 74.3 | 4 death | 11.4 | 1 SCD | |
| Ardan M. Saguner [23] | 2013 | 62 | 7.0 | 67.7 | 3 HTx/2 death | 8.1 | 7 SCA, 15 VF, 6 supraventricular arrhythmias | |
| Ardan M. Saguner [24] | 2014 | 70 | 5.3 | 67.1 | 5 HTx | 7.1 | 2 SCA, 25 sustained VT, 7 VF | |
| Jørg Saberniak [25] | 2014 | 110 | 58.2 | 5 HTx | 4.6 | 66 VA | ||
| Anneline S.J.M. te Riele [26] | 2015 | 75 | 9.0 | 54.7 | 2 HTx/2 death | 5.3 | 11 SCD, 8 SCA, 16 sustained VT | |
| Judith A. Groeneweg [14] | 2015 | 439 | 7.0 | 64.2 | 18 HTx/6 death | 5.8 | 21 SCA, 96 sustained VT, 38 atrial arrhythmias | |
| Aditya Bhonsale [11] | 2015 | 541 | 6.0 | 58.8 | 8 HTx/12 death | 3.7 | 36 SCD, 16 SCA | |
| Cristina Gallo [27] | 2016 | 68 | 1970–2014 | 17.0 | 69.1 | 3 HTx/4 death | 10.3 | 3 SCD, 1 SCA |
| Yoshitaka Kimura [28] | 2016 | 110 | 10.0 | 75.5 | 2 HTx/8 death | 9.1 | 74 VT/VF | |
| Aditya Bhonsale [29] | 2016 | 502 | 52.6 | 19 HTx/53 death | 14.3 | 34 SCD, 24 SCA, 167 sustained VT | ||
| Nisha A. Gilotra [10] | 2017 | 289 | 1998–2014 | 50.9 | 15 HTx/7 death | 7.6 | 2 SCD, 6 SCA | |
| Thomas Gilljam [30] | 2018 | 183 | 1988–2015 | 67.2 | 28 HTx | 15.3 | 45 VT, 8 SCA, 18 atrial arrhythmias | |
| Gabriela M. Orgeron [31] | 2017 | 312 | 7.0 | 52.2 | 2 HTx/12 death | 4.5 | 158 sustained VT, 19 VF | |
| Saagar Mahida [32] | 2019 | 110 | 2000–2015 | 6.4 | 82.7 | 10 HTx/3 death | 11.8 | 3 SCD |
| Annina S. Vischer [9] | 2019 | 135 | 7.0 | 61.5 | 5 HTx/3 death | 5.9 | - | |
| Erpeng Liang [33] | 2019 | 522 | 1995–2017 | 4.3 | 71.5 | 53 HTx/62 death | 22.0 | 14 SCD, 136 sustained VT |
| Shibu Mathew [34] | 2019 | 47 | 1998–2016 | 4.2 | 100 | 1 HTx/2 death | 6.4 | 4 SCA, 18 sustained VT |
| Mikael Laredo [35] | 2019 | 23 | 2003–2015 | 3.9 | 100 | 4 HTx | 17.4 | 19 VT |
| Alexis Hermida [36] | 2019 | 118 | 2006–2013 | 5.6 | 72.9 | 9 HTx/1 death | 8.5 | 54 SVT/VF/SCA/SCD |
| Elizabeth S. DeWitt [13] | 2019 | 32 | 56.3 | 10 HTx | 31.3 | 5 SCA, 14 VT | ||
| L. P. Bosman [37] | 2019 | 850 | 9.5 | 52.1 | 7 HTx/53 death | 7.1 | - |
3. Clinical Characteristics and Classification
The clinical course of HF in ACM was reported to be heterogeneous. Some ACM patients reached the endpoint of cardiac death or HTx within 2 or 3 years, while these occurred after a few decades in some patients [15][29][30]. The description of HF ’s course in ACM was limited in most studies and the potential relationship between HF and ventricular arrhythmias was incompletely described [38].
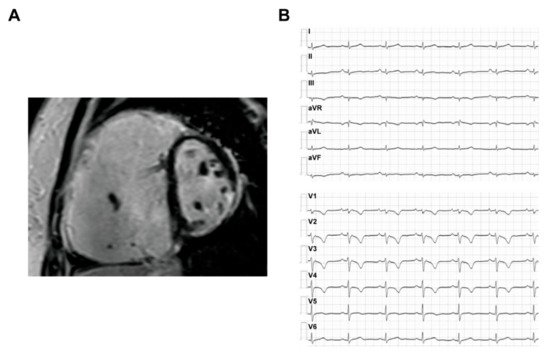
The ARVC patients could have a typical phenotype with both structural abnormalities and electrophysiological disorders, which are often reflected on the results of imaging and ECG tests. The typical CMR and ECG characteristics of ACM HF patients are provided in Figure 2 .
In a proteome study, heat shock protein 70 (HSP70) was significantly elevated in both ACM and other cardiomyopathies [40]. Furthermore, bridging integrator 1 (BIN1) [41][42], ST2 [43] and galectin-3 (GAL-3) [44] can reflect the severity of HF in ACM. The circulation level of complements is also correlated with mortality and cardiac dysfunction in ACM [45]. The sC5b6 level could display the severity of HF, which is significantly higher in ACM patients with biventricular dysfunction compared with isolated RV dysfunction. As is well-known, the abnormality of lipid metabolism is one of the key pathogenesis for ACM progression. The oxidized low-density lipoprotein (ox-LDL) could not only reflect the severity of ACM fat infiltration, but also predict the HF and malignant arrhythmia’s event risk. Another specific plasma biomarker for ACM clinical course discrimination and cardiac function prediction is β-hydroxybutyrate (β-OHB). In the report from Fuwai Hospital, the level of β-OHB is relatively low in healthy volunteers and unsuspected relatives of ACM patients. However, it is gradually increased in ACM patients following the clinical heart failure stage from normal cardiac function to isolated RV dysfunction and biventricular cardiac dysfunction. It could be recognized as a useful predictor for disease progression and adverse heart failure outcomes.
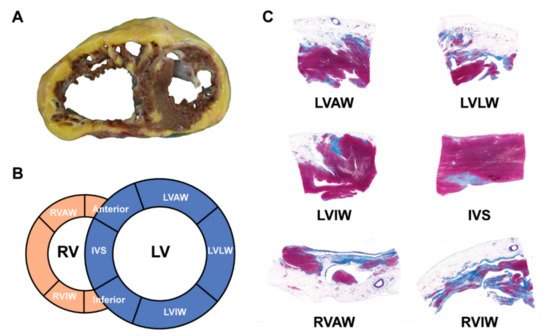
The hearts from orthotopic HTx and/or autopsies in ACM patients had distinctive histopathological features as compared to other cardiomyopathies. The typical gross morphology and histopathological characteristics of ARVC HF patients are provided in Figure 3 .
4. Risk Factors and Stratification
The average age of onset for ACM is 30–40. It is widely accepted that early-onset of HF often indicates more rapid occurrence of adverse outcomes. On the other hand, in a small cohort with young ACM patients under 18 years and with long-term follow-up, the incidence of adverse HF outcomes was similar in children and adults [26]. In the report from the Nordic ACM Registry, an age of disease onset under 35 years was shown to be an independent risk factor for HTx [30]. In contrast, older patients with late presentation of ACM, who were thought to be more vulnerable, had less risk of cardiac death and HTx than younger patients [29].
There is widespread consensus that family history is an important risk factor for ventricular arrhythmic events in ACM patients. ACM probands who had symptomatic relatives have a higher possibility for carrying desmosomal mutation and a higher risk of SCD [46]. With respect to the risk of HF, however, most reports showed that familial or isolated ACM probands had the same risk of developing HF [14][19][30]. Meanwhile, no evidence showed that ethnic background could affect the HF risk in ACM patients [10].
Arrhythmias may occur at any stage of ACM, and over 90% of patients had an initial manifestation or the history of VT, atrial arrhythmia or aborted SCD [13][47][48]. Many studies demonstrated the critical role of arrhythmias in the progression of HF. Theoretically, patients with recurrent VT and atrial arrhythmias tended to have worse hemodynamics features and pump dysfunction [23][32][38]. However, there was no evidence suggesting that VT is a risk factor for HF progression. In addition, the right bundle branch block (RBBB) was reported to have a significant correlation with severe biventricular HF [17]. The development of complete RBBB may lead to poor prognosis [49].
Several studies focused on the characteristics of atrial arrhythmias in ACM and found that these arrhythmias may be common, with a prevalence of about 10–20% in the general ACM population [19][30][38]. The incidence of atrial fibrillation in patients with adverse HF outcomes was 6–19% [30]. Atrial arrhythmias were associated with increasing mortality and morbidity and appeared to be risk factors for HTx and deaths due to end-stage HF. In addition, the incidence of first-degree atrioventricular block may be significantly higher in patients with rehospitalization for HF, and it was shown to be an independent risk factor for HF hospitalization in ACM [50].
This entry is adapted from the peer-reviewed paper 10.3390/jcm10204782
References
- Basso, C.; Corrado, D.; Marcus, F.I.; Nava, A.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009, 373, 1289–1300.
- Corrado, D.; Link, M.S.; Calkins, H. Arrhythmogenic Right Ventricular Cardiomyopathy. N. Engl. J. Med. 2017, 376, 61–72.
- Hoorntje, E.T.; Te Rijdt, W.P.T.; James, C.A.; Pilichou, K.; Basso, C.; Judge, D.P.; Bezzina, C.R.; Van Tintelen, J.P. Arrhythmogenic cardiomyopathy: Pathology, genetics, and concepts in pathogenesis. Cardiovasc. Res. 2017, 113, 1521–1531.
- Bosman, L.P.; Sammani, A.; James, C.A.; Cadrin-Tourigny, J.; Calkins, H.; van Tintelen, J.P.; Hauer, R.N.W.; Asselbergs, F.W.; Te Riele, A. Predicting arrhythmic risk in arrhythmogenic right ventricular cardiomyopathy: A systematic review and meta-analysis. Heart Rhythm 2018, 15, 1097–1107.
- Cadrin-Tourigny, J.; Bosman, L.P.; Nozza, A.; Wang, W.; Tadros, R.; Bhonsale, A.; Bourfiss, M.; Fortier, A.; Lie, O.H.; Saguner, A.M.; et al. A new prediction model for ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2019, 40, 1850–1858.
- Calkins, H.; Corrado, D.; Marcus, F. Risk Stratification in Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2017, 136, 2068–2082.
- Hauer, R.N.W. Prevention of Sudden Cardiac Death in Arrhythmogenic Cardiomyopathy. JACC Clin. Electrophysiol. 2018, 4, 769–770.
- Mazzanti, A.; Ng, K.; Faragli, A.; Maragna, R.; Chiodaroli, E.; Orphanou, N.; Monteforte, N.; Memmi, M.; Gambelli, P.; Novelli, V.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy: Clinical Course and Predictors of Arrhythmic Risk. J. Am. Coll. Cardiol. 2016, 68, 2540–2550.
- Vischer, A.S.; Castelletti, S.; Syrris, P.; McKenna, W.J.; Pantazis, A. Heart failure in patients with arrhythmogenic right ventricular cardiomyopathy: Genetic characteristics. Int. J. Cardiol. 2019, 286, 99–103.
- Gilotra, N.A.; Bhonsale, A.; James, C.A.; Te Riele, A.S.J.; Murray, B.; Tichnell, C.; Sawant, A.; Ong, C.S.; Judge, D.P.; Russell, S.D.; et al. Heart Failure Is Common and Under-Recognized in Patients With Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia. Circ. Heart Fail. 2017, 10.
- Bhonsale, A.; Groeneweg, J.A.; James, C.A.; Dooijes, D.; Tichnell, C.; Jongbloed, J.D.H.; Murray, B.; Te Riele, A.S.J.M.; Van Den Berg, M.P.; Bikker, H.; et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur. Heart J. 2015, 36, 847–855.
- Komura, M.; Suzuki, J.-I.; Adachi, S.; Takahashi, A.; Otomo, K.; Nitta, J.; Nishizaki, M.; Obayashi, T.; Nogami, A.; Satoh, Y.; et al. Clinical Course of Arrhythmogenic Right Ventricular Cardiomyopathy in the Era of Implantable Cardioverter-Defibrillators and Radiofrequency Catheter Ablation. Int. Heart J. 2010, 51, 34–40.
- DeWitt, E.S.; Chandler, S.F.; Hylind, R.J.; Beausejour Ladouceur, V.; Blume, E.D.; VanderPluym, C.; Powell, A.J.; Fynn-Thompson, F.; Roberts, A.E.; Sanders, S.P.; et al. Phenotypic Manifestations of Arrhythmogenic Cardiomyopathy in Children and Adolescents. J. Am. Coll. Cardiol. 2019, 74, 346–358.
- Groeneweg, J.A.; Bhonsale, A.; James, C.A.; Te Riele, A.S.; Dooijes, D.; Tichnell, C.; Murray, B.; Wiesfeld, A.C.; Sawant, A.C.; Kassamali, B.; et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ. Cardiovasc. Genet. 2015, 8, 437–446.
- Peters, S. Long-term follow-up and risk assessment of arrhythmogenic right ventricular dysplasia/cardiomyopathy: Personal experience from different primary and tertiary centres. J. Cardiovasc. Med. 2007, 8, 521–526.
- Peters, S.; Trümmel, M.; Meyners, W. Prevalence of right ventricular dysplasia-cardiomyopathy in a non-referral hospital. Int. J. Cardiol. 2004, 97, 499–501.
- Peters, S.; Peters, H.; Thierfelder, L. Heart failure in arrhythmogenic right ventricular dysplasia-cardiomyopathy. Int. J. Cardiol. 1999, 71, 251–256.
- Hulot, J.-S.; Jouven, X.; Empana, J.-P.; Frank, R.; Fontaine, G. Natural History and Risk Stratification of Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Circulation 2004, 110, 1879–1884.
- Lemola, K.; Brunckhorst, C.; Helfenstein, U.; Oechslin, E.; Jenni, R.; Duru, F. Predictors of adverse outcome in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy: Long term experience of a tertiary care centre. Heart 2005, 91, 1167–1172.
- Dalal, D.; Nasir, K.; Bomma, C.; Prakasa, K.; Tandri, H.; Piccini, J.; Roguin, A.; Tichnell, C.; James, C.; Russell, S.D.; et al. Arrhythmogenic right ventricular dysplasia: A United States experience. Circulation 2005, 112, 3823–3832.
- Watkins, D.A.; Hendricks, N.; Shaboodien, G.; Mbele, M.; Parker, M.; Vezi, B.Z.; Latib, A.; Chin, A.; Little, F.; Badri, M.; et al. Clinical features, survival experience, and profile of plakophylin-2 gene mutations in participants of the Arrhythmogenic Right Ventricular Cardiomyopathy Registry of South Africa. Heart Rhythm 2009, 6, S10–S17.
- Pinamonti, B.; Dragos, A.M.; Pyxaras, S.A.; Merlo, M.; Pivetta, A.; Barbati, G.; Di Lenarda, A.; Morgera, T.; Mestroni, L.; Sinagra, G. Prognostic predictors in arrhythmogenic right ventricular cardiomyopathy: Results from a 10-year registry. Eur. Heart J. 2011, 32, 1105–1113.
- Saguner, A.M.; Medeiros-Domingo, A.; Schwyzer, M.A.; On, C.-J.; Haegeli, L.M.; Wolber, T.; Hürlimann, D.; Steffel, J.; Krasniqi, N.; Rüeger, S.; et al. Usefulness of Inducible Ventricular Tachycardia to Predict Long-Term Adverse Outcomes in Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Cardiol. 2013, 111, 250–257.
- Saguner, A.M.; Vecchiati, A.; Baldinger, S.H.; Rüeger, S.; Medeiros-Domingo, A.; Mueller-Burri, A.S.; Haegeli, L.M.; Biaggi, P.; Manka, R.; Lüscher, T.F.; et al. Different prognostic value of functional right ventricular parameters in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ. Cardiovasc. Imaging 2014, 7, 230–239.
- Saberniak, J.; Hasselberg, N.E.; Borgquist, R.; Platonov, P.G.; Sarvari, S.I.; Smith, H.; Ribe, M.; Holst, A.G.; Edvardsen, T.; Haugaa, K.H. Vigorous physical activity impairs myocardial function in patients with arrhythmogenic right ventricular cardiomyopathy and in mutation positive family members. Eur. J. Heart Fail. 2014, 16, 1337–1344.
- Te Riele, A.S.J.M.; James, C.A.; Sawant, A.C.; Bhonsale, A.; Groeneweg, J.A.; Mast, T.P.; Murray, B.; Tichnell, C.; Dooijes, D.; Van Tintelen, J.P.; et al. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy in the Pediatric Population Clinical Characterization and Comparison with Adult-Onset Disease. JACC Clin. Electrophysiol. 2015, 1, 551–560.
- Gallo, C.; Blandino, A.; Giustetto, C.; Anselmino, M.; Castagno, D.; Richiardi, E.; Gaita, F. Arrhythmogenic right ventricular cardiomyopathy: ECG progression over time and correlation with long-term follow-up. J. Cardiovasc. Med. 2016, 17, 418–424.
- Kimura, Y.; Noda, T.; Otsuka, Y.; Wada, M.; Nakajima, I.; Ishibashi, K.; Miyamoto, K.; Okamura, H.; Aiba, T.; Kamakura, S.; et al. Potentially Lethal Ventricular Arrhythmias and Heart Failure in Arrhythmogenic Right Ventricular Cardiomyopathy: What Are the Differences Between Men and Women? JACC Clin. Electrophysiol. 2016, 2, 546–555.
- Bhonsale, A.; Te Riele, A.S.; Sawant, A.C.; Groeneweg, J.A.; James, C.A.; Murray, B.; Tichnell, C.; Mast, T.P.; van der Pols, M.J.; Cramer, M.J.; et al. Cardiac phenotype and long-term prognosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia patients with late presentation. Heart Rhythm 2017, 14, 883–891.
- Gilljam, T.; Haugaa, K.H.; Jensen, H.K.; Svensson, A.; Bundgaard, H.; Hansen, J.; Dellgren, G.; Gustafsson, F.; Eiskjær, H.; Andreassen, A.K.; et al. Heart transplantation in arrhythmogenic right ventricular cardiomyopathy—Experience from the Nordic ARVC Registry. Int. J. Cardiol. 2018, 250, 201–206.
- Orgeron, G.M.; James, C.A.; Riele, A.T.; Tichnell, C.; Murray, B.; Bhonsale, A.; Kamel, I.R.; Zimmerman, S.L.; Judge, D.P.; Crosson, J.; et al. Implantable Cardioverter-Defibrillator Therapy in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy: Predictors of Appropriate Therapy, Outcomes, and Complications. J. Am. Heart Assoc. 2017, 6.
- Mahida, S.; Venlet, J.; Saguner, A.M.; Kumar, S.; Baldinger, S.H.; AbdelWahab, A.; Tedrow, U.B.; Castelletti, S.; Pantazis, A.; John, R.M.; et al. Ablation compared with drug therapy for recurrent ventricular tachycardia in arrhythmogenic right ventricular cardiomyopathy: Results from a multicenter study. Heart Rhythm 2019, 16, 536–543.
- Liang, E.; Wu, L.; Fan, S.; Li, X.; Hu, F.; Zheng, L.; Fan, X.; Chen, G.; Ding, L.; Yao, Y. Bradyarrhythmias in Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Cardiol. 2019, 123, 1690–1695.
- Mathew, S.; Saguner, A.M.; Schenker, N.; Kaiser, L.; Zhang, P.; Yashuiro, Y.; Lemes, C.; Fink, T.; Maurer, T.; Santoro, F.; et al. Catheter Ablation of Ventricular Tachycardia in Patients With Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: A Sequential Approach. J. Am. Heart Assoc. 2019, 8, e010365.
- Laredo, M.; Da Silva, L.O.; Extramiana, F.; Lellouche, N.; Varlet, É.; Amet, D.; Algalarrondo, V.; Waintraub, X.; Duthoit, G.; Badenco, N.; et al. Catheter ablation of electrical storm in patients with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm 2020, 17, 41–48.
- Hermida, A.; Fressart, V.; Hidden-Lucet, F.; Donal, E.; Probst, V.; Deharo, J.C.; Chevalier, P.; Klug, D.; Mansencal, N.; Delacretaz, E.; et al. High risk of heart failure associated with desmoglein-2 mutations compared to plakophilin-2 mutations in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur. J. Heart Fail. 2019, 21, 792–800.
- Bosman, L.P.; Verstraelen, T.E.; van Lint, F.H.M.; Cox, M.; Groeneweg, J.A.; Mast, T.P.; van der Zwaag, P.A.; Volders, P.G.A.; Evertz, R.; Wong, L.; et al. The Netherlands Arrhythmogenic Cardiomyopathy Registry: Design and status update. Neth. Heart J. 2019, 27, 480–486.
- Camm, C.F.; James, C.A.; Tichnell, C.; Murray, B.; Bhonsale, A.; Te Riele, A.; Judge, D.P.; Tandri, H.; Calkins, H. Prevalence of atrial arrhythmias in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Heart Rhythm 2013, 10, 1661–1668.
- Chen, L.; Song, J.; Chen, X.; Chen, K.; Ren, J.; Zhang, N.; Rao, M.; Hu, Z.; Zhang, Y.; Gu, M.; et al. A novel genotype-based clinicopathology classification of arrhythmogenic cardiomyopathy provides novel insights into disease progression. Eur. Heart J. 2019, 40, 1690–1703.
- Wei, Y.-J.; Huang, Y.-X.; Shen, Y.; Cui, C.-J.; Zhang, X.-L.; Zhang, H.; Hu, S.-S. Proteomic analysis reveals significant elevation of heat shock protein 70 in patients with chronic heart failure due to arrhythmogenic right ventricular cardiomyopathy. Mol. Cell. Biochem. 2009, 332, 103–111.
- Asimaki, A. BIN1: A new biomarker to track ARVC? Heart Rhythm 2012, 9, 968–969.
- Hong, T.-T.; Cogswell, R.; James, C.A.; Kang, G.; Pullinger, C.R.; Malloy, M.J.; Kane, J.P.; Wojciak, J.; Calkins, H.; Scheinman, M.M.; et al. Plasma BIN1 correlates with heart failure and predicts arrhythmia in patients with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm 2012, 9, 961–967.
- Broch, K.; Leren, I.S.; Saberniak, J.; Ueland, T.; Edvardsen, T.; Gullestad, L.; Haugaa, K.H. Soluble ST2 is associated with disease severity in arrhythmogenic right ventricular cardiomyopathy. Biomarkers 2017, 22, 367–371.
- Oz, F.; Onur, I.; Elitok, A.; Ademoglu, E.; Altun, I.; Bilge, A.K.; Adalet, K. Galectin-3 correlates with arrhythmogenic right ventricular cardiomyopathy and predicts the risk of ventricular arrhythmias in patients with implantable defibrillators. Acta Cardiol. 2017, 72, 453–459.
- Ren, J.; Tsilafakis, K.; Chen, L.; Lekkos, K.; Kostavasili, I.; Varela, A.; Cokkinos, D.V.; Davos, C.H.; Sun, X.; Song, J.; et al. Crosstalk between coagulation and complement activation promotes cardiac dysfunction in arrhythmogenic right ventricular cardiomyopathy. Theranostics 2021, 11, 5939–5954.
- Caforio, A.L.P.; Re, F.; Avella, A.; Marcolongo, R.; Baratta, P.; Seguso, M.; Gallo, N.; Plebani, M.; Izquierdo-Bajo, A.; Cheng, C.Y.; et al. Evidence From Family Studies for Autoimmunity in Arrhythmogenic Right Ventricular Cardiomyopathy: Associations of Circulating Anti-Heart and Anti-Intercalated Disk Autoantibodies With Disease Severity and Family History. Circulation 2020, 141, 1238–1248.
- Groeneweg, J.A.; van der Zwaag, P.A.; Olde Nordkamp, L.R.; Bikker, H.; Jongbloed, J.D.; Jongbloed, R.; Wiesfeld, A.C.; Cox, M.G.; van der Heijden, J.F.; Atsma, D.E.; et al. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy According to Revised 2010 Task Force Criteria With Inclusion of Non-Desmosomal Phospholamban Mutation Carriers. Am. J. Cardiol. 2013, 112, 1197–1206.
- Link, M.S.; Laidlaw, D.; Polonsky, B.; Zareba, W.; McNitt, S.; Gear, K.; Marcus, F.; Estes, N.A., 3rd. Ventricular arrhythmias in the North American multidisciplinary study of ARVC: Predictors, characteristics, and treatment. J. Am. Coll. Cardiol. 2014, 64, 119–125.
- Peters, S.; Trümmel, M.; Koehler, B. Special features of right bundle branch block in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Int. J. Cardiol. 2012, 157, 102–103.
- Kimura, Y.; Noda, T.; Matsuyama, T.-A.; Otsuka, Y.; Kamakura, T.; Wada, M.; Ishibashi, K.; Inoue, Y.; Miyamoto, K.; Okamura, H.; et al. Heart failure in patients with arrhythmogenic right ventricular cardiomyopathy: What are the risk factors? Int. J. Cardiol. 2017, 241, 288–294.