Peptides are increasingly being developed for use as therapeutics to treat many ailments, including cancer. Therapeutic peptides have the advantages of target specificity and low toxicity. The anticancer effects of a peptide can be the direct result of the peptide binding its intended target, or the peptide may be conjugated to a chemotherapy drug or radionuclide and used to target the agent to cancer cells. Peptides can be targeted to proteins on the cell surface, where the peptide–protein interaction can initiate internalization of the complex, or the peptide can be designed to directly cross the cell membrane. Peptides can induce cell death by numerous mechanisms including membrane disruption and subsequent necrosis, apoptosis, tumor angiogenesis inhibition, immune regulation, disruption of cell signaling pathways, cell cycle regulation, DNA repair pathways, or cell death pathways.
- drug delivery
- peptide therapeutic
- covalent-based peptide inhibitors
- PCNA
1. Introduction
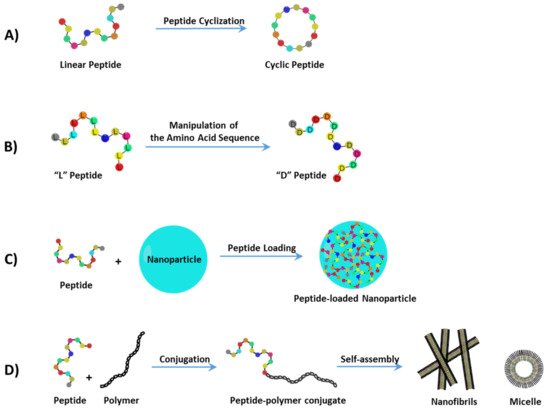
2. Treating Cancer with Cell-Targeting Peptide (CTP) and Cell-Permeable Peptide (CPP)
2.1. Cell-Targeting Peptides
2.2. Cell-Penetrating Peptides
3. Possible Mechanisms of Therapeutic Peptides
4. Rational of Targeting PCNA-Binding Proteins with a Peptide Derived from Cancer-Associated PCNA
The development of new peptide stability and delivery strategies is opening novel avenues for designing peptide therapeutics that can target key proteins in tumorigenesis. Proliferating cell nuclear antigen (PCNA) is one such target, and it is an essential protein involved in many processes including DNA replication, DNA repair, chromatin organization, transcription, sister chromatin cohesion and cell cycle control [81,82]. PCNA is highly expressed in cancer cells and is critical for cellular proliferation.
A common strategy of designing peptide-based therapy is to disrupt protein–protein interactions (PPIs) with known protein binding sites. PCNA is a homotrimer that encircles DNA and acts as a platform that binds and coordinate proteins at the replication fork [89,90]. It is thought to interact and regulate over a hundred proteins [91] with various functions in the cell. Many, but not all, interacting proteins have conserved motifs that bind to PCNA, such as PIP-box (PCNA-interacting protein box), APIM (AlkB homologue 2 PCNA-interacting motif), and KA box (consisting of residues K-A-(A/L/I)-(A/L/Q)-x-x-(L/V)) [92,93]. The interdomain connector loop (IDCL) and a proximal hydrophobic patch on PCNA is one region where proteins have been indicated to interact [94,95], such as the PIP-box of p21 [96], peptide from the p66 subunit of DNA polymerase δ [94], and FEN1 [97].
To design a therapeutic peptide from PCNA that is specifically cytotoxic toward malignant cells, early studies focused on how PCNA in breast cancer cells was different. An isoform of PCNA was found in malignant breast epithelial cells and tissues but not non-malignant cells, using 2-dimensional PAGE experiments [98]. The cancer-associated isoform of PCNA (caPCNA) was likely from post-translational modifications [99] and not from genetic mutations or alternate splicing [100]. Cells from prostate cancer, hepatic carcinoma, high-grade prostatic intraepithelial neoplasia, and neuroblastoma [101–103] also had the unique PCNA isoform associated with cancer. Antibodies were developed using peptides derived from PCNA and the antibodies were screened for their ability to recognize the caPCNA isoform. Epitope screening studies led to the discovery of an 8-amino acid peptide, dubbed caPeptide, within the IDCL. Conjugating nine D-arginines linked by two cysteines to the N-terminus of the peptide led to the development of a cell-permeable peptide, R9-caPeptide, that was found to selectively inhibit malignant cancer growth instead of non-malignant and normal cells [104,105]. R9-caPeptide is an example of a cationic CPP for delivery to cancer cells.
R9-caPeptide was found to disrupt interaction of PCNA with binding partners and growth inhibition experiments showed that R9-caPeptide was cytotoxic in a dose-dependent manner to cancer cell lines derived from breast, lymphoma, neuroblastoma, and pancreas [88,104,105,107]. Treating malignant cancer cells with R9-caPeptide also caused stalled DNA replication forks, DNA damage, cell cycle arrest, and apoptosis. Validating the therapeutic potential, R9-caPeptide inhibited, in mice, xenograft tumor growth from triple negative breast cancer and from neuroblastoma cell lines [104,105].
5. Use of Covalent Warheads in Peptide-Based Therapeutics
Peptide-based therapeutics may provide a viable means to target PPIs, such as for PCNA, as perturbing interactions between partners that involve large surface areas can be a significant challenge for organic small molecule-based therapies. Peptides inhibitors to PPIs can be generated from peptide library screening strategies, or from structural studies that have characterized the PPI interface. Peptides identified by these approaches typically require further optimization of their drug-likeness through improving: affinity, selectivity, stability and cell permeability. An interesting direction to improve affinity and selectivity has been to add functional groups that can form covalent interactions with the protein target side chains in the binding site; such covalent coupling to the protein target provides a greatly enhanced potency of the inhibitor. Acrylamides and chloroacetamides have been extensively studied as moieties for targeting the cysteine side chain, and are being used in covalent targeting strategies for organic small molecules and peptide-based therapies. However, the ‘cysteinome’, proteins containing targetable cysteine residues, is somewhat limited due to the relatively rare occurrence of cysteine in a protein sequence. Thus, recent studies have been exploring potential chemistries to target other side chains and thereby extend the number of proteins that can be targeted through covalent-based approaches.
The discovery that aryl-sulfonyl fluorides and aryl-fluoro sulfates can act as covalent-warheads within peptide inhibitors notably expands the list of covalently targetable residues to now include lysine, tyrosine or histidine side chains. Aryl-fluoro sulfates may be of the strongest interest for therapeutic development, as they were confirmed to be cell permeable and stable in both aqueous buffer and plasma [110]. However, a concern from an initial characterization of this covalent warhead was an observed slow reaction rate, questioning the overall effectiveness of this warhead in forming covalent adducts within the cell. The initial study targeted a lysine residue that was relatively distant from the protein binding site, and encouragingly, a follow up study instead targeted a lysine within the peptide binding site, and rapid covalent adduct formation was readily observed [111]. Similar results were also observed in a separate study targeting human Mcl-1, using a BH3 substrate peptide for generating pro-apoptotic agent [112]. Thus, the rapid bond formation together with the cellular permeability, and stability being akin to that previously observed with acrylamides targeting cysteine, further suggests that aryl-fluoro sulfates may well expand the targetable residues beyond the cysteinome for novel therapeutic development. Other forms of potential covalent modifications strategies may be focused on the peptide substrate itself, adding stability and structure, to improve its binding characteristics. Cyclization of the peptide has been a common approach in this regard, and more recent studies have suggested other methods, including N-locking. A lactam bond is formed between the amino terminus and a glutamic residue at position 4, and this N-lock can nucleate helix formation within the peptide. N-locking can also be coupled with the covalent warhead strategy, as observed in BH3 peptides that were developed to target the antiapoptotic protein Bfl-1 to produce a peptide that was soluble in aqueous buffer and had low nanomolar affinity to its target [113]. Thus, these covalent-based approaches may provide novel avenues to develop peptide compounds with potentially more suitable ADME characteristics, and higher affinities and activities against their cellular targets.
6. Conclusion
A major consideration in developing peptide therapeutics is addressing delivery problems that prevent adequate quantities of peptide to reach cancer cells. To increase selectivity to malignant tissues and decrease toxicity, peptides can carry a load, such as a chemotherapy agent or another peptide that binds to proteins involved in tumorigenesis. Not only can peptide-based drugs disrupt essential PPIs utilized during the progression of cancer growth, but they can also disrupt membranes, affect the vascularization of the tumor, or induce an immune response that leads to cell death. The strategies and development of peptide therapeutics described here show promise in the laboratory with potential application to future cancer treatments.
This entry is adapted from the peer-reviewed paper 10.3390/cells10112908