Alzheimer’s disease (AD) is the most frequently diagnosed neurodegenerative disorder worldwide and the sixth leading cause of death in the United States and is on the rise [1]. The disorder is characterised by amyloid plaques and neurofibrillary tangles; cell loss, vascular damage and dementia follow as a direct result of the vicious cycle of their deposition. Along with age and family history, inheritance also plays an essential role in the development of AD. Among several genetic risk factors, the APP, PSEN1, PSEN2 had been identified as a causative factor for early-onset AD, the APOE-ε4 allele (encodes a protein that transports cholesterol in the bloodstream) was shown to have a strong impact on late-onset AD. However, studies based on APOE status among different racial and ethnic groups have shown inconsistent results .Despite a number of treatments being approved by the U.S. Food and Drug Administration (FDA), none of those therapeutic strategies can cure the disease . There is strong evidence that early diagnosis and treatment might help to decelerate the progression of the disease and maintain brain function. Therefore, rational development of medical approaches (e.g., sophisticated brain imaging studies and discovery of novel candidate genes, proteins, and other substances in blood or cerebrospinal fluid) are fundamental for better understanding of the molecular factors that contribute to disease progression, and for improving the early diagnosis and treatment decisions.
- Alzheimer’s disease
- gene co-expression network
- genome-scale metabolic model
1. Distinct Gene Expression and Functional Profiles in Different Brain Regions of AD
| VS | p-Val (Hyper) | Overlap | Region_1 DEG Size |
Region_2 DEG Size |
Both CodingGene Size |
|---|---|---|---|---|---|
| RO-MT | 0.00054 | 128 | 2885 | 477 | 14,001 |
| MC-RO | 0.00516 | 347 | 2885 | 1515 | 14,186 |
| MT-MC | 9.20599 × 10−11 | 98 | 477 | 1515 | 14,186 |
| Gene Symbol | Associated Protein Name (UNIPROT) | Most Associated KEGG Pathways |
|---|---|---|
| ADAMTS2 ¥ | A disintegrin and metalloproteinase with thrombospondin motifs 2 | - |
| ATP1B3 # | Sodium/potassium-transporting ATPase subunit beta-3 | Secretion (insulin, salivary, bile, gastric acid, pancreatic, aldosterone) has04961_Endocrine_and_other_factor-regulated_calcium_reabsorption has04973_Carbohydrate_digestion_and_absorption has04260_Cardiac_muscle_contraction has04978_Mineral_absorption |
| BTG # | B-cell translocation gene 1 protein | has03018_RNA_degradation |
| DAXX # | Death domain-associated protein 6 | has04010_MAPK_signaling_pathway, has04210_Apoptosis, has05012_Parkinson_disease, has05014_Amyotrophic_lateral_sclerosis, has05022_Pathways_of_neurodegeneration, has05168_Herpes_simplex_virus_1_infection |
| DYNC2LI1 * | Cytoplasmic dynein 2 light intermediate chain 1 | has04962_Vasopressin-regulated_water_reabsorption, has05132_Salmonella_infection |
| FAM129B # | Protein Niban 2 | - |
| FAM167B # | Protein FAM167B | - |
| FAM90A1 * | Protein FAM90A1 | - |
| FBXO2 # | F-box only protein 2 | has04068_FoxO_signaling_pathway, has04120_Ubiquitin_mediated_proteolysis, has04141_Protein_processing_in_endoplasmic_reticulum, has05132_Salmonella_infection |
| GARNL3 # | GTPase-activating Rap/Ran-GAP domain-like protein 3 | - |
| GIT1 # | ARF GTPase-activating protein GIT1 | has04144_Endocytosis, has04810_Regulation_of_actin_cytoskeleton, has05120_Epithelial_cell_signaling_in_Helicobacter_pylori_infection |
| H2AFV # | Histone H2A.V | - |
| HEBP2 # | Heme-binding protein 2 | - |
| ID3 # | DNA-binding protein inhibitor ID-3 | has04350_TGF-beta_signaling_pathway |
| NHEJ1 # | Non-homologous end-joining factor 1 | has03450_Non-homologous_end-joining |
| NT5DC2 # | 5’-nucleotidase domain-containing protein 2 | - |
| PAFAH1B3 # | Platelet-activating factor acetylhydrolase IB subunit alpha1 | has00565_Ether_lipid_metabolism |
| PLAGL1 * | Zinc finger protein PLAGL1 | - |
| RAF1 # | RAF proto-oncogene serine/threonine protein kinase | CENTRAL Signaling (JAK/STAT, TNF, VEGF, Insulin, Apelin, cAMP, mTOR etc.) Cancer (colorectal, pancreatic, breast, glioma, melanoma etc.) Infection (Hepatitis, Influenza, Tuberculosis, Salmonella etc.) has04210_Apoptosis, has04218_Cellular_senescence, has04510_Focal_adhesion, has04540_Gap_junction, has04935_Growth_hormone_synthesis, secretion_and_action has04726_Serotonergic_synapse, has04510_Focal_adhesion |
| RALBP1 # | RalBP1-associated Eps domain-containing protein 2 | has04014_Ras_signaling_pathway, has05212_Pancreatic_cancer |
| S100A4 # | Calvasculin/Metastasin | - |
| S100A6 # | Calcyclin/Growth factor-inducible protein 2A9 | - |
| SEPT9 # | Septin-9 | - |
| SMAD4 # | Mothers against decapentaplegic homolog 4 | Signalling (Fox0, Wnt, apelin etc.), Cancer (colorectal etc.) has04110_Cell_cycle, has04520_Adherens_junction |
| STARD10 # | START domain-containing protein 10 | - |
| STAT5B # | Signal transducer and activator of transcription 5B | Signalling (AGE-RAGE, JAK/STAT etc.), Myeloid leukemia has04217_Necroptosis, has04659_Th17_cell_differentiation |
| TRIM45 * | Tripartite motif-containing protein 45 | - |
| TRIM66 * | Tripartite motif-containing protein 66 | - |
| TRIP10 # | Cdc42-interacting protein 4 | has04910_Insulin_signaling_pathway |
| TTC14 * | Tetratricopeptide repeat protein 14 | - |
| UBAP1 # | Ubiquitin-associated protein 1 | - |
| UBXN8 * | UBX domain-containing protein 8 | has04141_Protein_processing_in_endoplasmic_reticulum |
| ZNF334 * | Zinc finger protein 334 | has05168_Herpes_simplex_virus_1_infection |
| ZNF639 # | Zinc finger protein 636 | - |
| Pathway | Shared DEGs in the Pathway | |||
|---|---|---|---|---|
| hsa03450_Non-homologous_end-joining | NHEJ1 | |||
| hsa04010_MAPK_signaling_pathway | DAXX | RAF1 | ||
| hsa04012_ErbB_signaling_pathway | RAF1 | STAT5B | ||
| hsa04014_Ras_signaling_pathway | RAF1 | RALBP1 | ||
| hsa04015_Rap1_signaling_pathway | RAF1 | |||
| hsa04022_cGMP-PKG_signaling_pathway | ATP1B3 | RAF1 | ||
| hsa04024_cAMP_signaling_pathway | ATP1B3 | RAF1 | ||
| hsa04062_Chemokine_signaling_pathway | RAF1 | STAT5B | ||
| hsa04068_FoxO_signaling_pathway | RAF1 | SMAD4 | ||
| hsa04071_Sphingolipid_signaling_pathway | RAF1 | |||
| hsa04072_Phospholipase_D_signaling_pathway | RAF1 | |||
| hsa04150_mTOR_signaling_pathway | RAF1 | |||
| hsa04151_PI3K-Akt_signaling_pathway | RAF1 | |||
| hsa04261_Adrenergic_signaling_in_cardiomyocytes | ATP1B3 | |||
| hsa04210_Apoptosis | DAXX | RAF1 | ||
| hsa04310_Wnt_signaling_pathway | SMAD4 | |||
| hsa04350_TGF-beta_signaling_pathway | ID3 | SMAD4 | ||
| hsa04370_VEGF_signaling_pathway | RAF1 | |||
| hsa04371_Apelin_signaling_pathway | RAF1 | SMAD4 | ||
| hsa04390_Hippo_signaling_pathway | SMAD4 | |||
| hsa04550_Signaling_pathways_regulating_pluripotency_of_stem_cells | RAF1 | ID3 | SMAD4 | |
| hsa04625_C-type_lectin_receptor_signaling_pathway | RAF1 | |||
| hsa04630_JAK-STAT_signaling_pathway | RAF1 | STAT5B | ||
| hsa04659_Th17_cell_differentiation | SMAD4 | STAT5B | ||
| hsa04660_T_cell_receptor_signaling_pathway | RAF1 | |||
| hsa04662_B_cell_receptor_signaling_pathway | RAF1 | |||
| hsa04664_Fc_epsilon_RI_signaling_pathway | RAF1 | |||
| hsa04722_Neurotrophin_signaling_pathway | RAF1 | |||
| hsa04910_Insulin_signaling_pathway | RAF1 | TRIP10 | ||
| hsa04912_GnRH_signaling_pathway | RAF1 | |||
| hsa04915_Estrogen_signaling_pathway | RAF1 | |||
| hsa04917_Prolactin_signaling_pathway | RAF1 | STAT5B | ||
| hsa04919_Thyroid_hormone_signaling_pathway | RAF1 | ATP1B3 | ||
| hsa04921_Oxytocin_signaling_pathway | RAF1 | |||
| hsa04926_Relaxin_signaling_pathway | RAF1 | |||
| hsa04933_AGE-RAGE_signaling_pathway_in_diabetic_complications | SMAD4 | STAT5B | ||
| hsa04935_Growth_hormone_synthesis,_secretion_and_action | RAF1 | STAT5B | ||
| hsa05120_Epithelial_cell_signaling_in_Helicobacter_pylori_infection | GIT1 | |||
| hsa05161_Hepatitis_B | RAF1 | SMAD4 | STAT5B | |
| hsa05200_Pathways_in_cancer | RAF1 | RALBP1 | SMAD4 | STAT5B |
| hsa05210_Colorectal_cancer | RAF1 | SMAD4 | ||
| hsa05212_Pancreatic_cancer | RAF1 | RALBP1 | SMAD4 | |
| hsa05213_Endometrial_cancer | RAF1 | |||
| hsa05215_Prostate_cancer | RAF1 | |||
| hsa05219_Bladder_cancer | RAF1 | |||
| hsa05220_Chronic_myeloid_leukemia | RAF1 | SMAD4 | STAT5B | |
| hsa05221_Acute_myeloid_leukemia | RAF1 | STAT5B | ||
| hsa05223_Non-small_cell_lung_cancer | RAF1 | STAT5B | ||
| hsa05224_Breast_cancer | RAF1 | |||
| hsa05226_Gastric_cancer | RAF1 | SMAD4 | ||
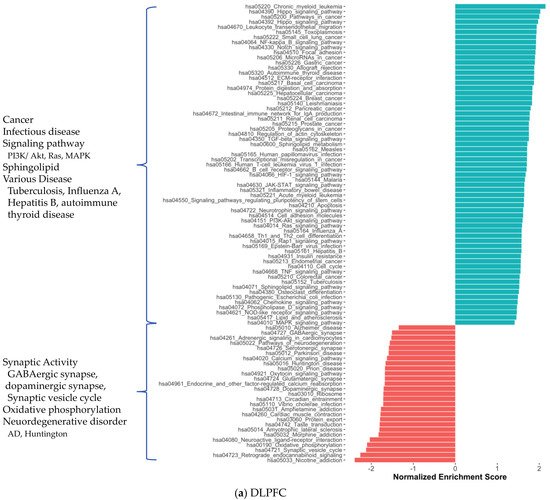
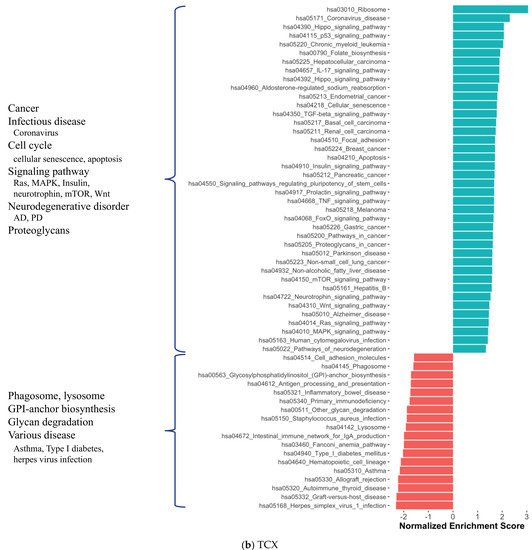
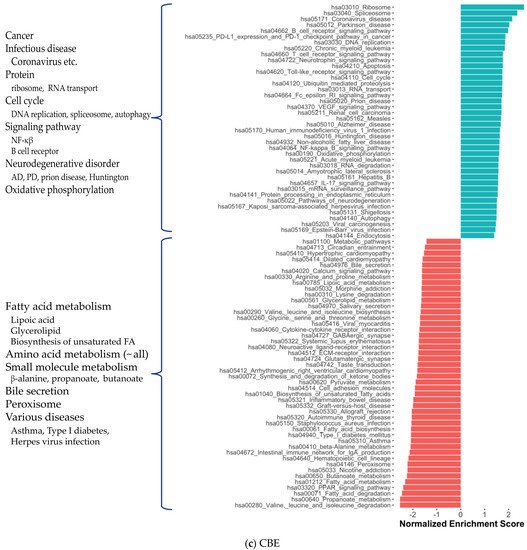
2. Co-Expression Network Analysis in Different Brain Regions of AD
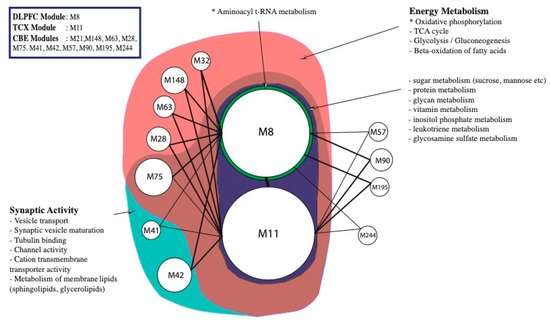
3. Alterations in Energy Metabolism, Chaperones and Synaptic Activity
| Tissues | Hub Genes (Top 10%) |
|---|---|
| DLPFC, TCX, CBE | AMIGO1, GPRASP2 |
| DLPFC, TCX | Cytoskeleton and its organisation ACTR3B, GABARAPL1, MARK1, NDEL1 Synaptic activity/plasticity AP2M1, ARHGEF9, CALM3, DLG3, GABRB3, L1CAM, NPTN Intra Golgi and retrograde Golgi-to-ER traffic AP3M2, ARF3, CFAP36, KIFAP3, KLC1, NAPB, NSF, RAB6B Ubiquitin/Proteasome System DNAJC5, HSPA12A Glucose Metabolism/Oxidative phosphorylation ATP6V1A, ATP6V1B2, HK1, SEH1L, SLC25A14, SLC25A4, SLC9A6 Other ATL1, B4GAT1, BTRC, C1orf216, CDK14, CDK5R1CHN1, CISD1, CLSTN3, CNTNAP1, EID2, FAM234B, FAM49A, GLS, GOT1, GPI, GUCY1B3, INPP4A, JAZF1, MAGEE1, MAPK9, MLTT11, MOAP1, MYCBP2, NDFIBP1, NDRG3, NELL2, NMNAT2, OPCML, PCMT1, PFN2, PHACTR1, PNMA2, PNMAL1, PPPIR7, PPP3CB, PPP3R1, PREP, PREPL, PRKCE, RBFOX2, REEP1, REEP5, RTN3, SEPT6, SMAP2, SNAP91, SV2B, SYT13, TMEM246, TSPYL1, UBE2O, UBFD1, VDAC1, VDAC3, WDR7, YWHAG, YWHAZ |
| TCX, CBE | Cytoskeleton and its organisation PAK1 |
| DLPFC, CBE | Glucose Metabolism/Oxidative phosphorylation ATP6V1E1 Cytoskeleton and its organisation CDC42, DCTN2 Other ERLEC1, MPP1 |
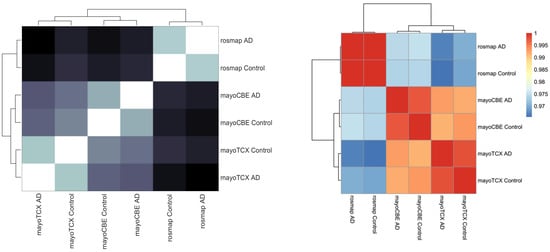
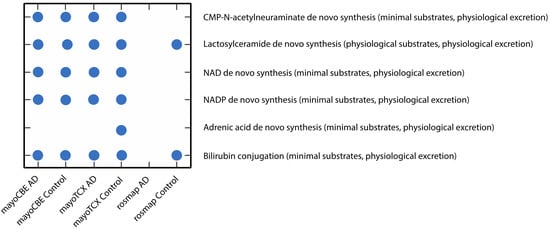
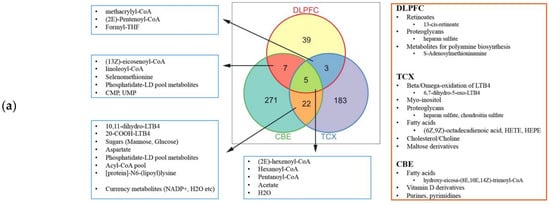
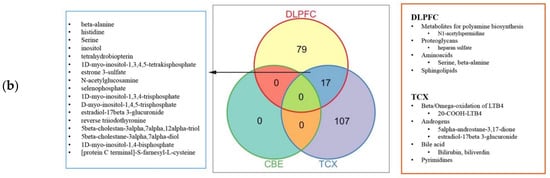
2.4. Metabolic Alterations in Different Brain Regions of AD

This entry is adapted from the peer-reviewed paper 10.3390/ijms222111556