Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Neurosciences
NRF2 (Nuclear Factor-Erythroid 2 Like 2) is a transcription factor that orchestrates the cellular response to oxidative stress. The regulation of NRF2 signalling has been shown to be a promising strategy to modulate the progression of the neurodegeneration associated to Parkinson’s disease. The NRF2 pathway has been shown to be affected in patients with this disease, and activation of NRF2 has neuroprotective effects in preclinical models, demonstrating the therapeutic potential of this pathway.
- NRF2
- Parkinson’s disease
1. Dopaminergic Neurons as Vulnerable Targets of Oxidative Stress
Dopaminergic neurons in the SN have several traits that make them especially sensitive to oxidative stress [19,20]: Anatomically, dopaminergic neurons have a long-range neuronal projection with complex dendritic and axonal arborization, coupled with glutamatergic innervation from the subthalamic nucleus, and a high microglia concentration was observed in the SN. Metabolically, dopaminergic neurons are very active, with an autonomous pacemaking activity that requires high oxygen consumption, a finely tuned calcium signalling process, and cellular proteostasis. Dopaminergic neurons in the SN exhibit elevated rates of oxidative phosphorylation in the mitochondria, resulting not only in higher ATP production, but also in ROS generation compared to neighbouring cells in the ventral tegmental area. Additionally, dopamine metabolism, the presence of iron, and low levels of antioxidants make them particularly prone to oxidative stress. Oxidative stress is not only present throughout PD, but has also been detected in early disease stages [21], suggesting an important role in the pathogenesis of the disease.
Dopamine itself is a source of ROS (Figure 1) that can damage dopaminergic neurons [22]. Dopamine generated in the cytoplasm or uptaken by the dopamine transporter present in dopaminergic neurons is autoxidized, inducing an increase in dopamine quinones that are toxic to the cells. Neurotoxins 6-hydroxydopamine and tetrahydroisoquinoline alkaloids can be produced by a non-enzymatic reaction involving dopamine, H2O2, and free iron, all of them present in dopaminergic neurons [23].
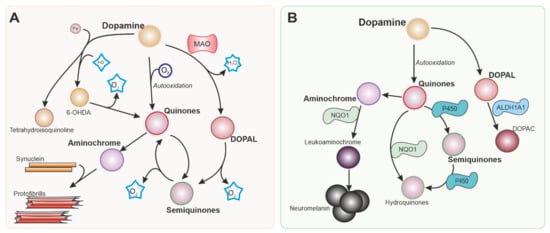
Figure 1. Intracellular dopamine metabolism. Parkinson’s disease is characterized by the degeneration of dopaminergic neurons in the substantia nigra. Dopamine metabolism is responsible in part for the vulnerability of the neurons in this nucleus. Reactions generating reactive oxygen species (ROS) from dopamine and its metabolites are shown in (A). Nrf-2 regulates several enzymes represented in (B) involved in detoxifying reactions that can mitigate the production of ROS and metabolites that are toxic for dopaminergic neurons. MAO: Monoamine oxidase. 6-OHDA: 6-hydroxydopamine. NQO1: NADPH quinone dehydrogenase 1. ALDH1A: Aldehyde dehydrogenase 1.
Monoamine oxidases (MAO) are enzymes that generate H2O2 as a by-product of the metabolism of catecholamines and indoleamines. While MAO-B is located primarily in glial cells, MAO-A is present in neurons [24], including dopaminergic neurons in the SN, although at relatively low levels [25]. MAO-B is expressed at high levels in astrocytes in the SN, and the H2O2 produced by this enzyme can cross the cell membrane and affect neighbouring cells, as well as promote excitotoxicity [26].
NOXs are source of ROS, with a special importance in phagocytic cells. Upregulation and release of ROS are hallmarks of activated microglia [27]. Activation of NOX led to an increased microglial activation and dopaminergic cell death in cultures [15]. We demonstrated that NOX activation can increase ROS levels and decrease cell survival also in pure neuronal cultures [28].
2. Antioxidant Defences and NRF2
Cells have evolved several antioxidant mechanisms to counteract the effect of ROS and avoid damage. Nonenzymatic ROS scavengers include vitamins and their precursors that have direct scavenging effect on ROS and are obtained mostly by food intake. Other molecules with ROS scavenging activity (NADPH, uric acid, glutathione, taurine, thioredoxin…) can, on the contrary, be regenerated by the activity of cellular enzymes.
Enzymes that react to electrophilic chemicals and xenobiotics had been classified as phase I, phase II, and phase III depending on their function. NRF2 is a transcription factor that regulates gene expression of phase I, II, and III enzymes responsible of antioxidant defence [29]. Under normal circumstances, NRF2 is sequestered in the cytoplasm by KEAP1 (Kelch-like ECH-associated protein 1), an inhibitor of its function that facilitates its degradation by ubiquitination [30,31]. In the presence of ROS, reactive nitrogen species, and electrophilic compounds, KEAP1 is modified causing the dissociation of KEAP1 from NRF2, allowing the stabilization of NRF2 and its translocation and accumulation in the nucleus. Alternatively, kinase-mediated phosphorylation of KEAP1 can also induce KEAP1 inactivation and NRF2 translocation to the nucleus [29,32]. Once in the nucleus, NRF2 can bind to the promoter regions of genes containing “antioxidant response element” sequences, promoting the expression of antioxidant genes, including NRF2 itself [33].
Dopaminergic neurons are not only exposed to different sources of ROS, but also have compromised antioxidant defence mechanisms. Depending on the source and type of ROS, cells use combinations of ROS scavengers and enzymes that maintain the redox potential of the cell. NRF2 translocates to the nuclei of dopaminergic neurons and increases the transcription of target genes such as Heme Oxygenase 1 (HMOX1) and NAD(P)H Quinone dehydrogenase 1 (NQO1), which are found in the brains of patients with idiopathic PD [34,35,36]. Besides NRF2 activation in the SN of patients with PD, systemic activation of the NRF2 pathway has been recently reported [37,38,39,40], thus this pathway has been proposed as a marker of PD. In dopaminergic neurons, NRF2 regulates genes that mediate dopamine metabolism (Figure 1B) and various antioxidant systems (Figure 2).
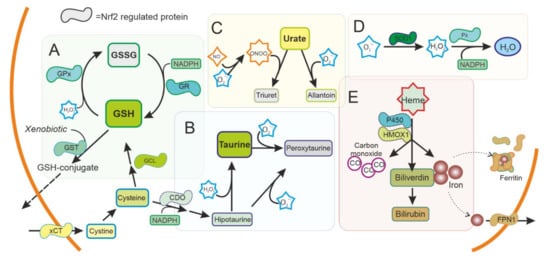
Figure 2. Antioxidant system regulated by NRF2 in the cytoplasm. ROS production is used as a signal molecule and is a byproduct of several reactions in cells that can result in oxidative stress. Cells can use ROS scavengers such as (A) glutathione (GSH), (B) taurine, (C) urate, as well as detoxifying enzymes as (D) superoxide dismutase (SOD), peroxidase (Px), (E) Heme oxygenase 1 (HMOX1), or cytochrome P450, present in the cytoplasm to reduce ROS and toxic metabolites. NRF2 is regulating and can be regulated by these pathways. GSH: Reduced glutathione. GSSG oxidized glutathione. GST: Glutathione S Transferase. GPx: Glutathione peroxidase. GR: Glutathione reductase. xCT: Cystine-Glutamate exchanger. GCL Glutamate-cysteine ligase. CDO Cysteine dioxygenase. SOD1: Superoxide dismutase type 1. FPN1: Ferroportin.
Disorders affecting GSH metabolism are common in major neurodegenerative diseases. In human brains, the levels of GSH peroxidase correlate with the survival of dopaminergic neurons in PD [41] and reduced glutathione levels have been found in the brain of patients with PD [42]. NRF2 regulates the levels of not only GSH peroxidase, but most other key enzymes for GSH synthesis and regeneration (Cystine/glutamate antiporter (xCT), γ-glutamate cysteine ligase (GCL) subunits, glutathione reductase (GR), glutathione S-transferases (GSTs), and others) (Figure 2A). These enzymes have been shown to be affected also in PD brains [43]. Besides GSH peroxidase, there are many other enzymes with peroxidase activity in the thioredoxins superfamily of enzymes that also participate in the control of redox signalling [44]. Most of them are regulated by NRF2, and together function as a signalling system that regulates NRF2 pathway and consequently their own expression [45,46].
Taurine (2-aminoethanesulfonic acid) is the most abundant intracellular amino acid from a very early age in humans, with levels particularly high in excitable tissues that are susceptible to oxidative stress, such as the brain. A reduction in taurine levels has been shown in patients with PD and other neurodegenerative disorders that correlates with the progression of the disease [47]. Taurine can act as an antioxidant and have neuroprotective effects [48,49]. Mechanistically, taurine protects neuronal cells by decreasing superoxide generation from mitochondria, reducing the damage to more sensitive antioxidant systems and, indirectly, by decreasing microglia activation and the oxidative stress associated to microglial NADPH-derived ROS that cause damage in neuronal cells [49,50,51,52]. NRF2 promotes the synthesis of taurine at the expense of cysteine and NADPH, possibly affecting other antioxidant systems, and conversely taurine increases the expression of NRF2 and downstream genes [53,54].
NADH and NADPH are cofactors essential for maintaining cellular redox homeostasis by providing reducing equivalents to antioxidant enzymes. These molecules are obtained in the tricarboxylic acid cycle in the mitochondria and the pentose phosphate pathway in the cytoplasm. NRF2 is key in regulating the production of these cofactors [55,56], most notably by regulating the pentose phosphate pathway, and tricarboxylic acid cycle intermediates are able to activate the NRF2 pathway [57,58].
Urate is an antioxidant that can scavenge peroxynitrite and hydroxyl radical. Interestingly, urate is the end product of purine metabolism in humans because of the absence of a functional urate oxidase gene. Urate oxidase is present in animal models, and its disruption has been shown to protect dopaminergic cells both in vivo and in vitro [59]. Urate levels are lower in patients with PD [60] and this affects NRF2 expression regulating the antioxidant and inflammatory response [61].
SOD enzymes are also able to reduce oxidative stress by eliminating superoxide. SOD1, also called CuZnSOD, is located in the cytosol, mitochondrial intermembrane space, and peroxysomes (Figure 3); SOD2 or MnSOD is located mainly in the mitochondria, while SOD3 is mainly extracellular. These enzymes have been shown to be regulated by NRF2 [62], and have been linked to PD [63].
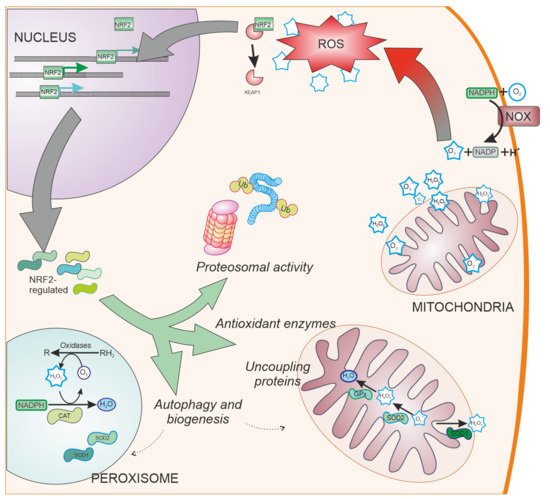
Figure 3. NRF2-regulated systems. Besides ROS scavengers located in the cytoplasm, organelles involved in oxygen metabolism such as mitochondria and peroxisomes, contain NRF2-regulated antioxidant enzymes specialized in the antioxidant metabolism such as catalase (CAT), superoxide dismutase type 1 and type 2 (SOD1 and SOD2, respectively), and peroxidases. Additionally, NRF2 regulates other cell pathways involved in reducing oxidative stress production, such as elimination of proteins by proteosomal degradation or autophagy and organelle biogenesis. NOX: NADPH oxidase. Ub: Ubiquitin. CAT: Catalase.
NRF2 is well known to regulate two genes that also have antioxidant activity, HMOX1 and NQO1. The products of these genes metabolize heme and quinone, molecules that can generate ROS, and thus both have been classified as “detoxifying enzymes” [29].
HMOX1 is an inducible enzyme responsible for heme degradation, resulting in carbon monoxide, free iron, and biliverdin (Figure 2E). HMOX1 expression was upregulated in glial cells in animal models of PD [64] and has been found upregulated in the SN of patients with PD, both in surviving dopaminergic neurons and astrocytes [35]. HMOX1 activity has been associated with cytoprotective effects, although the neuroprotective effect of HMOX1 has been questioned [65,66]. HMOX1 has been shown to have an anti-inflammatory effect and switches macrophages from proinflammatory to anti-inflammatory phenotype [67], suggesting an important role in regulating microglia in PD. Other enzymes related to iron and heme metabolism, including ferritin and ferroportin, are also regulated by NRF2 [68,69]. Levels of ferritin have been found to be decreased in the SN of patients with PD [70]. Ferritin sequesters free iron in microglia with a neuroprotective effect [71].
NQO1 is implicated in the detoxification of quinones (Figure 1B). This is important in dopaminergic neurons since dopamine and other catecholamines can autoxidize to form quinones that can be toxic for dopaminergic neurons before polymerizing to form neuromelanin. 6-hydroxydopamine (6-OHDA), the first dopaminergic neurotoxin discovered, has been also shown to cause dopaminergic neuronal death via quinone formation [72]. Both dopamine and 6-OHDA have been shown to activate the NRF2 pathway and NQO1 [73,74]. NQO1 is localized in dopaminergic neurons in the SN and ventral tegmental area [75]. Elevated NQO1 levels were found in patients with PD [33,76], but NQO1 immunoreactivity is virtually absent when dopaminergic neurons degenerate in advanced stages of the disease [36]. NQO1 has been suggested to protect against several insults associated with PD [77,78]. However, similarly to HMOX1, the neuroprotective effects of NQO1 have been questioned [79].
Another gene of interest for PD is CYP2D6. This gene codifies cytochrome P450, a phase I enzyme induced by NRF2 that is highly expressed in liver and brain, where it is involved in drug metabolism. In the brain, it was found expressed at high levels in the SN [80,81], where it is located in dopaminergic neurons [82]. Cytochrome P450 has been shown to have neuroprotective effect in models of PD [83]. Polymorphisms of this enzyme have been associated with PD risk [84] and its expression is decreased in patients with PD compared to age-matched controls [85].
Cells have evolved other pathways to reduce oxidative stress by decreasing ROS generation (Figure 3). These pathways include the ubiquitin proteasome system [86], uncoupling mitochondrial proteins [87], and organelle autophagy [88] or biogenesis through PGC-1α [89] and PPARγ [90]. NRF2 is involved in the regulation of these pathways [91,92,93], having shown neuroprotective effects in models of PD [94,95].
3. Oxidative Stress in Familial Forms of PD: Relationship with NRF2
Oxidative stress is one of the possible mechanisms involved in the pathogenesis and progression of idiopathic forms of PD. Besides those, there are a few genes that have been linked with the disease. For a long time, these genes have been associated with redox imbalance, and their relation with NRF2 is becoming apparent over the more recent years [96].
Synuclein (SNCA) is a major component of the Lewy body, one of the hallmarks of PD, and mutations and even overexpression of the wild type SNCA cause familial forms of the disease. Misfolded synuclein causes microglial activation and increased expression of antioxidant response enzymes [97], suggesting a role in regulating NRF2 pathway. NRF2 expression has been shown to be neuroprotective in cellular and animal models expressing α-synuclein [98,99]. In cellular models, downregulation of NRF2 and HMOX1 induces synuclein aggregation [100]. Mutant synuclein causes mitochondrial dysfunction and an increase in ROS levels, while NRF2 activation can reduce oxidative stress and ameliorate mitochondrial damage [99].
Mutations in the PARK2 gene parkin (PRKN) show impaired ubiquitin protein ligase activity. Cells use parkin to target proteins to be degraded by the UPS and reduce oxidative and endoplasmic reticulum stress [101]. In induced pluripotent stem cells (iPSC)-derived neurons, mutant PRKN decreased levels of GSH, increased levels of ROS production, and elevated NRF2 and NQO1. Defects in mitochondria were detected in these neurons, but not in undifferentiated iPSCs nor the fibroblast from patients from which these cells were derived [76].
PINK1 is a protein located in the mitochondrial membrane that can interact with parkin. Together, they regulate mitochondria maintenance by sensing damaged mitochondria. PINK1 has neuroprotective properties by labelling defective mitochondria for selective degradation via autophagy (also called “mitophagy”) [102]. NRF2 can directly upregulate PINK1 in response to oxidative stress [103] and PINK1 reduce mitochondria-derived ROS overproduction by inducing mitophagy. NRF2 activity is upregulated by autophagy, promoting the expression of p62 and PGC-1α, which are key regulators of the recycling of mitochondria and lysosomes [10,104].
DJ1 gene encodes a highly conserved, ubiquitous protein with functions that are not so well-known. An antioxidant role of DJ1 has been proposed early due to its association with PD [105,106], with prominent expression in glial cells and upregulation of GSH synthesis in PD models [107]. Even before being recognized as a gene responsible for familial forms of PD, DJ1 was found to be oxidized in response to toxins used in PD modelling and microglial activation [108]. DJ-1 directly regulates NRF2 by associating with KEAP1, thus avoiding NRF2 degradation and facilitating its nuclear accumulation [109]. The interaction between DJ-1 and NRF2 is well known and is considered key for the role of DJ-1 in PD pathogenesis [110].
LRRK2 (Leucine-Rich Repeat Kinase 2) is one of the most prevalent genes associated with familial forms of PD. Wild type LRRK2 expression increases cell survival in oxidative stress conditions in culture, while viability was decreased in cells carrying a mutant form of LRRK2 [111]. In individuals carrying the LRRK2 mutation but without PD, the levels of urate (an NRF2 activator, see above) were higher than in affected patients with the same mutation, suggesting that urate has a protective role and can be used as a biomarker of resistance to PD [112]. An association between NRF2 concentration and UPDRS scores was found in PD carriers of LRRK2 mutations [113], but there are no statistically significant differences between the levels of NRF2 in CSF of patients with PD with LRRK2 mutations compared to healthy LRRK2 carriers.
PARK5 gene, UCHL-1 (Ubiquitin Carboxyl-terminal Hydrolase L1), is a deubiquitinating enzyme that is affected by oxidative stress in PD [114]. Although association studies between NRF2 and UCHL-1 in PD have not yet been carried out, UCHL-1 is co-regulated with NRF2 in hyperglycaemia models [115], and NRF2 pathway has been proposed as a possible therapeutic approach for traumatic brain injury, where UCHL-1 is also upregulated [116]; however, a direct link between these two remains to be established. Other genes associated with PD have shown some association with NRF2, but further research is required to stablish NRF2 relevance to other familial forms of PD.
The enzymes and proteins involved in cell protection against oxidative stress, as well as genes associated with familial forms of PD, are, in many cases, present in astrocytes or microglial cells, and not necessarily in neurons: GSH formation requires the interplay between different cells, neuromelanin accumulated inside dopaminergic neurons can be released and activate microglial cells, and strong evidence supports non-cell autonomous degeneration in PD [117,118], including the evidence of cell to cell propagation of synuclein and fibrillary tangles [119]. The role of NRF2 in glial cells is discussed in the next section.
4. Involvement of Glial Cells in NRF2 Protection
PD affects primarily dopaminergic neurons, but many of the effects of the disease are mediated by glial cells. Many of the neuroprotective effects previously discussed are directly mediated by astrocytes or microglia [120,121,122]. Glial cells typically express higher levels of NRF2 [121,123] and this correlates with higher expression and more variety of antioxidant genes compared with neurons [21,35]. Both astrocytes an microglia express high levels of HMOX1 in early stages of the PD [98], and the role of glial cells has been studied extensively by Cuadrado’s research group in different models of the disease [124,125,126]. In PD, glutamate from the subthalamic nucleus can cause excitotoxicity, but high levels of glutamate also cause the inhibition of the import of cystine from astrocytes, resulting in reduced glutathione levels and a form of cell injury called oxidative glutamate toxicity or oxytosis [127]. Dopamine can activate the NRF2 pathway in astrocytes [128] and activation of the NRF2 in these cells supports the survival of dopaminergic neurons [129,130]. Dopamine also activates NRF2 and promotes iron accumulation in macrophages [131], and efficient iron homeostasis in microglial cells is protective in PD models [132]. Astrocytes can also store iron and reduce iron burden in neurons [133,134], as well as act as glutamate sinks. These mechanisms of iron accumulation are important in ferroptosis, an iron-dependent form of cell death closely related to oxytosis [127,135].
Astrocytic NRF2 is neuroprotective in animal models of PD [136], and the absence of NRF2 in astrocytes could impair the neuroprotection conferred to neurons expressing NRF2 [137]. Given the supporting role that astroglial cells have on neurons, astrocytes are ideal targets to direct therapeutic interventions involving the NRF2 pathway.
This entry is adapted from the peer-reviewed paper 10.3390/antiox10111649
This entry is offline, you can click here to edit this entry!