Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Health Care Sciences & Services
l-Carnitine is an amino acid derivative widely known for its involvement in the transport of long-chain fatty acids into the mitochondrial matrix, where fatty acid oxidation occurs. Moreover, l-Carnitine protects the cell from acyl-CoA accretion through the generation of acylcarnitines.
- l-carnitine
- fatty acid oxidation
1. Introduction
Carnitine (3-hydroxy-4-N-trimethylaminobutyrate) represents an amino acid derivative and a micronutrient that plays a key role in intermediary metabolism with the main function being the transport of long-chain fatty acids from the cytosol to the mitochondrial matrix where fatty acid β-oxidation occurs. Other established functions of carnitine are the preservation of membrane integrity [1], the stabilization of a physiologic coenzyme A (CoASH)/acetyl-CoA ratio in mitochondria, and the reduction of lactate production [2][3].
Carnitine is present in most, if not all, animal species and in several micro-organisms and plants. In the human body, carnitine is mainly found in a free form (free carnitine) and in the form of acylcarnitine esters, a pool of carnitine bound to various acyl groups that are delivered throughout the body for a wide range of functions [4]. At rest, the skeletal muscle carnitine pool is distributed as 80–90% free carnitine, 10–20% short-chain acylcarnitines (with a number of carbon atoms <10), and <5% long-chain acylcarnitines (with a number of carbon atoms >10) [5].
It has been estimated that the total carnitine content in the human body is about 300 mg/kg, with about 95% stored intracellularly in the heart and skeletal muscle, and the remaining part in the liver, kidney, and plasma [6]. The amount of circulating plasma carnitine accounts for only 0.5% of total body carnitine [7].
Carnitine does not undergo metabolic changes and, therefore, is eliminated as free carnitine in urine. However, a part of carnitine that is not absorbed at the level of the small intestine is completely degraded by bacteria in the large intestine to produce trimethylamine, a quaternary amine that, after enterocyte absorption, is oxidized in the liver by flavin-containing monooxygenase 3 to form trimethylamine-N-oxide (TMAO) [8].
In light of the fundamental role of carnitine in fatty acids β-oxidation for energy production, studies have been conducted to understand whether carnitine supplementation can affect skeletal muscle function and athletic performance in healthy individuals [2][9]. There is controversy as to whether or not carnitine administration can improve physical performance. The difference in exercise intensity, the training or conditioning of the subjects, the amount of carnitine administered, the route of administration, and the timing of administration relative to the exercise led to different experimental results [10].
2. Endogenous Synthesis and Cell Transport of l-Carnitine
Humans obtain carnitine mainly from the diet, predominantly from animal-based food products such as red meat, chicken, fish, and dairy. Only 25% of carnitine comes from endogenous synthesis [2][11]. Carnitine biosynthesis requires two essential amino acids: l-lysine, which provides the carbon backbone, and l-methionine that furnishes the N-methyl group [12]. The pathway of carnitine synthesis requires vitamin C, vitamin B6, niacin, and reduced iron as cofactors [13][14]. Lysine residues in some proteins undergo N-methylation using S-adenosylmethionine as a methyl donor, forming 6-N-trimethyl-lysine residues which are converted in carnitine in four enzymatic steps, namely hydroxylation at carbon 3, aldol cleavage, oxidation of the aldehyde to 4-butyrobetaine and hydroxylation of 4-butyrobetaine at carbon 3 [15]. The last enzymatic step is catalyzed by the enzyme 4-butyrobetaine dioxygenase to yield carnitine. It is generally recognized that the first three reactions of carnitine synthesis are widely distributed in the body but the final reaction is only present in the liver, kidney, and brain [7]. Thus, other tissues depend on carnitine uptake from the circulation.
Carnitine homeostasis is guaranteed by intestinal absorption from the diet, modest endogenous synthesis, and efficient renal reabsorption. Intestinal absorption of carnitine occurs by both passive (small intestine and colon) and active (duodenum and ileum) mechanisms [16]. The renal process ensures tubular reabsorption of about 98–99% of filtered carnitine thus conserving a normal carnitine level also in strict vegetarians (vegans) and lacto-ovo-vegetarians [17]. The renal threshold for carnitine excretion is about 50 µmol/L that is about the same value as the normal carnitine plasma concentration [18]. Thus, when plasma carnitine concentration raises, the renal excretion increases whereas the reabsorption decreases, thus maintaining baseline carnitine plasma level [18]. The opposite occurs when the plasma carnitine level is decreased, as in the case of low dietary intake of carnitine [18]. Neither renal reabsorption nor changes in dietary carnitine intake appear to affect the rate of endogenous carnitine synthesis [19][20].
The plasma membrane transport of carnitine is made by the carnitine/organic cation transporters (OCTN), a family of transporters that consists of three isoforms, i.e., OCTN1 (Scarnitine22A4) and OCTN2 (Scarnitine22A5) in humans and animals [21], and Octn3 (Slc22a21) in mice and humans [22]. Among them, OCTN2 is a high-affinity plasma membrane protein that can transport carnitine in a sodium-dependent manner and whose functional defect causes primary systemic carnitine deficiency [23]. OCTN2 is mainly involved in maintaining carnitine homeostasis that results from intestinal absorption, distribution to tissues, and renal excretion/reabsorption. OCTN2 has a high affinity for carnitine and its derivatives and it is present not only at the level of polarized cells of intestine, kidney, placenta and mammary gland but also in other tissues such as liver, heart, testis, skeletal muscle and brain [24][25]. OCTN1, highly expressed at the renal epithelium level and, to a lesser extent, in other tissues, is also involved in carnitine transport but with a lower affinity with respect to OCTN2 [23][24]. Moreover, other transporters such as ATB0,+ [26] and carnitine transporter-2 [27] can also carry carnitine.
Among different peripheral tissues, muscle is probably the main target of carnitine transport. About 90–95% of total carnitine is concentrated in muscle, and the ratio between muscle carnitine concentration and plasma concentration is around 50:1 [28].
Carnitine transport defect, associated with mutations in the OCTN2 transporter, causes profound intracellular and plasma carnitine depletion and very high renal carnitine excretion [29]. The consequence of such a defect is the inability of carnitine to perform its functions, particularly in β-oxidation. Metabolic and clinical aberrations associated with the defect in carnitine transport can be prevented by oral supplementation of pharmacological carnitine dosages (100–400 mg/kg/day). Due to the defective OCTN2, this treatment can increase the plasma carnitine level nearly to normal values but does not bring muscle carnitine concentration to normal values [29].
3. Role of Carnitine in Mitochondrial Fatty Acid Transport and β-Oxidation
Fatty acids from different sources (diet, endogenous de novo synthesis and adipose tissue hydrolysis) can cross the plasma membrane and enter the muscle cell by fatty acid transport proteins (FATPs), fatty acid translocase (FAT/CD36), caveolins and plasma membrane fatty acid-binding proteins (FABPpm) [30]. Once in the cell, long-chain fatty acids are activated to fatty acyl-CoAs by a family of acyl-CoA synthetases (ACS) identified at the plasma membrane, mitochondria, and lipid droplets [31]. FATPs have also ACS activity [32]. The activation reaction, which requires ATP and CoASH, traps long-chain fatty acyl-CoA thioesters inside the cell and, maintaining intracellular free long-chain fatty acids to low concentrations, allows further fatty acid uptake.
As long-chain acyl-CoA derivatives cannot directly cross the mitochondrial inner membrane, the entry of acyl-CoAs into the mitochondrial matrix for β-oxidation is guaranteed, under normal conditions, by carnitine palmitoyltransferase-1 (CPT-1), located on the external surface of the outer mitochondrial membrane [33]. CPT-1 catalyzes the transfer of acyl groups from acyl-CoA to carnitine to produce acylcarnitines and free CoASH (Figure 1). The reaction catalyzed by CPT-1 is tightly regulated to control both fatty acid β-oxidation and ketone body formation [34][35]. There are three different isoforms of CPT-1: CPT-1A, CPT-1B, and CPT-1C [35]. CPT-1 A is expressed in the liver, brain, kidney, lung, spleen, intestine, pancreas, ovary, and fibroblasts [36]. CPT-1B is the muscle isoform and is highly expressed in skeletal muscle, heart, and testis [36]. CPT1-C is the neuron-specific isoform, but its function in neural metabolism remains controversial [37]. CPT-1 is sensitive to inhibition by malonyl-CoA [38]. Since malonyl-CoA represents the product of acetyl-CoA carboxylase, a key enzyme of the cytosolic fatty acid synthesis pathway, malonyl-CoA can reciprocally regulate fatty acid synthesis and oxidation [38]. Thus, a high rate of fatty acid synthesis results in a low rate of fatty acid oxidation, and vice versa.
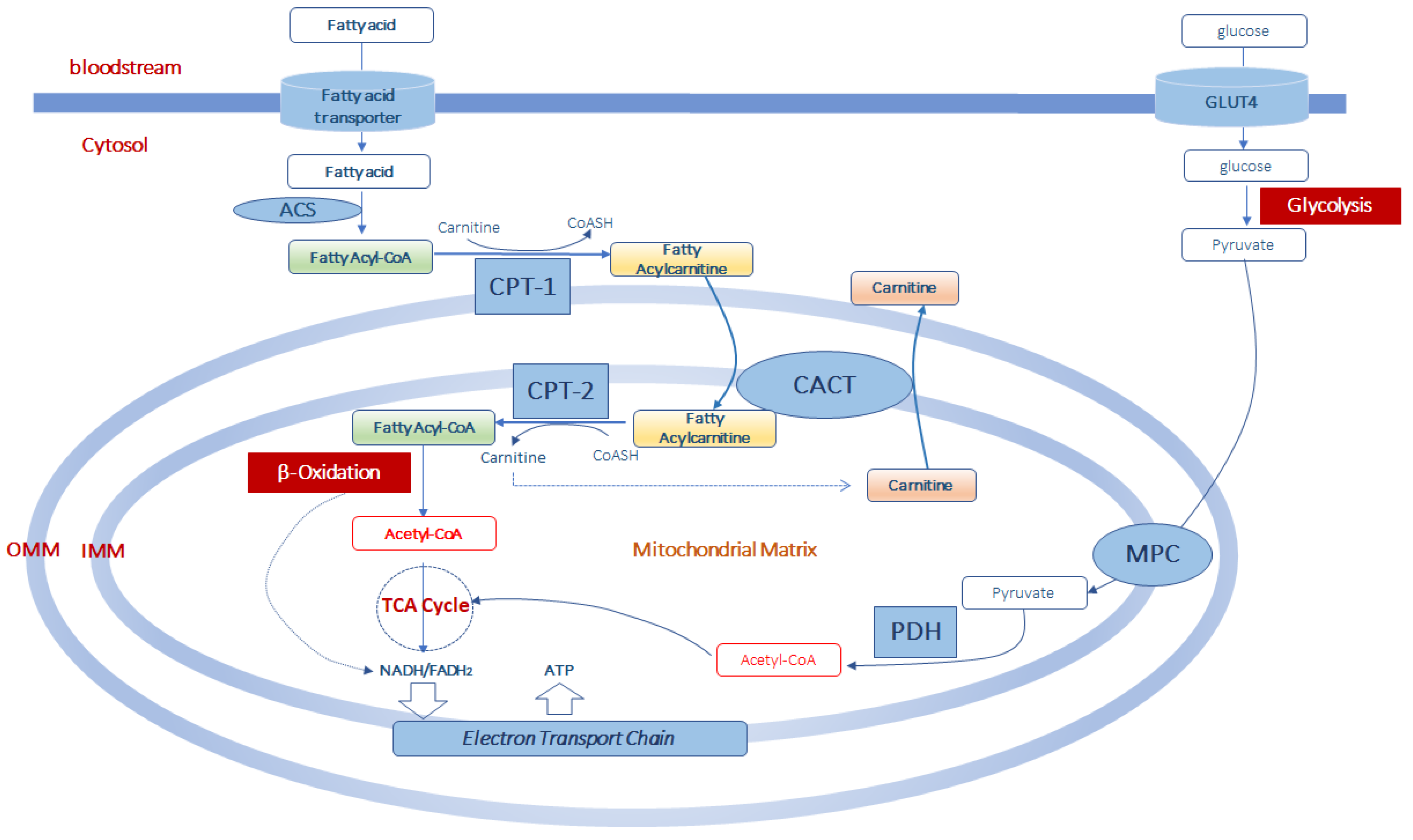
Figure 1. Acetyl-CoA production from fatty acid and glucose metabolism in muscle mitochondria. Fatty acids enter the cells by fatty acid transporters. Once in the cell they are activated to fatty acyl-CoA by acyl-CoA synthetase (ACS) before entering the mitochondria. At the level of the outer part of the outer mitochondrial membrane (OMM), fatty acyl-CoAs are bind to carnitine, by carnitine palmitoyltransferase-1 (CPT-1) activity, to form fatty acylcarnitine derivatives which diffuse through the outer mitochondrial membrane. Thus, the formed fatty acylcarnitines are transported across the inner mitochondrial membrane (IMM) via carnitine-acylcarnitine translocase (CACT). In the mitochondrial matrix, CPT-2 converts fatty acylcarnitines back to fatty acyl-CoAs, which enter the β-oxidation pathway and to free carnitine which can exit from the mitochondria in exchange with other acylcarnitines through CACT. Mitochondrial acetyl-CoA is generated from both β-oxidation of fatty acids and from pyruvate. Pyruvate is formed in the glycolytic pathway from glucose which enters the muscle cell via the glucose transporter type 4 (GLUT4). Pyruvate is transported in the mitochondrial matrix by the pyruvate carrier (MPC) of the inner mitochondrial membrane. Once in the mitochondria, pyruvate is converted into acetyl-CoA throughout the complex of the pyruvate dehydrogenase (PDH). This acetyl-CoA, together with that formed in the β-oxidation pathway can enter the tricarboxylic acid cycle (TCA cycle) to produce equivalent donors in the form of NADH (H+) and FADH2 which are oxidized in the mitochondrial electron transport chain to produce ATP.
Once synthesized, acylcarnitines cross the outer mitochondrial membrane, which is permeable to small molecules [39], and translocate into the matrix by the carnitine/acylcarnitine transporter (CACT), a protein of the inner mitochondrial membrane belonging to the mitochondrial carrier protein family [40][41]. CACT transports acylcarnitines in the matrix in exchange for intramitochondrial free carnitine [41][42][43]. Once in the mitochondrial matrix, fatty acyl units are transferred from carnitine to CoASH by carnitine palmitoyltransferase-2 (CPT-2) to form acyl-CoAs that enter the β-oxidation pathway (Figure 1).
The fatty acid β-oxidation, a mitochondrial process regulated by both nutritional and hormonal factors [44][45], involves the repetitive removal of two carbon units, in the form of acetyl-CoA, from the fatty acyl-chain. The enzymes 2-enoyl-CoA hydratase, 3-hydroxy acyl-CoA dehydrogenase, and 3-oxoacyl-CoA thiolase are subsequently involved in the fatty acid β-oxidation process to complete the conversion of the acyl-CoA ester into acetyl-CoA. The last step releases the two-carbon acetyl-CoA and a ready primed acyl-CoA that takes another turn down the spiral. In total each turn of the β-oxidation spiral produces reduced cofactors in the form of NADH (H+) and FADH2 and one acetyl-CoA. Further oxidation of acetyl-CoA via the Krebs cycle produces ATP and additional NADH (H+) and FADH2 [46]. Electrons from NADH (H+) and FADH2 pass through the electron transport chain, localized at the inner mitochondrial membrane, to oxygen which is reduced to water. During the passage of electrons throughout the different complexes of the electron transport chain is released energy to generate a transmembrane proton gradient which is used to generate ATP (Figure 1). The complete oxidation of a fatty acid produces numerous ATP molecules.
While the transport of long-chain acyl-CoAs, such as palmitoyl-CoA, oleoyl-CoA and linoleoyl-CoA, requires the presence of both CPT-1 and CPT-2, the transport and oxidation of medium- (C6-C12) and short-chain (C4-C6) fatty acids seems largely independent from the carnitine shuttle [47][48].
The hydroxyl group of carnitine can also form esters with acetate, to generate acetyl-carnitine, or with different carboxylic acids, including fatty acids of all chain lengths, to form a wide array of acylcarnitines [49].
This entry is adapted from the peer-reviewed paper 10.3390/molecules25010182
References
- Siliprandi, N.; Di Lisa, F.; Menabo, R. Clinical use of carnitine. Past, present and future. Adv. Exp. Med. Biol. 1990, 272, 175.
- Karlic, H.; Lohninger, A. Supplementation of l-carnitine in athletes: Does it make sense? Nutrition 2004, 20, 709–715.
- Friolet, R.; Hoppeler, H.; Krähenbühl, S. Relationship between the coenzyme A and the carnitine pools in human skeletal muscle at rest and after exhaustive exercise under normoxic and acutely hypoxic conditions. J. Clin. Investig. 1994, 94, 1490–1495.
- Reuter, S.E.; Evans, A.M. Carnitine and acylcarnitines: Pharmacokinetic, pharmacological and clinical aspects. Clin. Pharmacokinet. 2012, 51, 553–572.
- Hiatt, W.R.; Regensteiner, J.G.; Wolfel, E.E.; Ruff, L.; Brass, E.P. Carnitine and acylcarnitine metabolism during exercise in humans. Dependence on skeletal muscle metabolic state. J. Clin. Investig. 1989, 84, 1167–1173.
- Brass, E.P. Pharmacokinetic considerations for the therapeutic use of carnitine in hemodialysis patients. Clin. Ther. 1995, 17, 176–185.
- Flanagan, J.L.; Simmons, P.A.; Vehige, J.; Willcox, M.D.; Garrett, Q. Role of carnitine in disease. Nutr. Metab. 2010, 7, 30.
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585.
- Brass, E.P. Carnitine and sports medicine: Use or abuse? Ann. N. Y. Acad. Sci. 2004, 1033, 67–78.
- Heinonen, O.J. Carnitine and physical exercise. Sports Med. 1996, 22, 109–132.
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine transport and fatty acid oxidation. Biochim. Biophys. Acta 2016, 1863, 2422–2435.
- Tanphaichitr, V.; Broquist, H.P. Role of lysine and e-N-trimethyllysine in carnitine biosynthesis. Ii. Studies in the rat. J. Biol. Chem. 1973, 248, 2176–2181.
- Vaz, F.M.; Wanders, R.J. Carnitine biosynthesis in mammals. Biochem. J. 2002, 361, 417–429.
- Borum, P.R. Carnitine. Annu. Rev. Nutr. 1983, 3, 233–259.
- Adeva-Andany, M.M.; Calvo-Castro, I.; Fernández-Fernández, C.; Donapetry-García, C.; Pedre-Piñeiro, A.M. Significance of l-carnitine for human health. IUBMB Life 2017, 69, 578–594.
- Hamilton, J.W.; Li, B.U.; Shug, A.L.; Olsen, W.A. Carnitine transport in human intestinal biopsy specimens. Demonstration of an active transport system. Gastroenterology 1986, 91, 10–16.
- Rebouche, C.J. Kinetics, pharmacokinetics, and regulation of l-carnitine and acetyl-l-carnitine metabolism. Ann. N. Y. Acad. Sci. 2004, 1033, 30–41.
- Evans, A.M.; Fornasini, G. Pharmacokinetics of L-carnitine. Clin. Pharmacokinet. 2003, 42, 941–967.
- Rebouche, C. Carnitine: Modern Nutrition in Health and Disease; Shils, M., Shike, M., Ross, A., Eds.; Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2006; pp. 537–544.
- Mohammed, A.; Majid, A.; Ayman, W.E. Carnitine inborn errors of metabolism. Molecules 2019, 24, 3251.
- Tamai, I. Pharmacological and pathophysiological roles of carnitine/organic cation transporters (OCTNs: Scarnitine22A4, Scarnitine22A5 and Slc22a21). Biopharm. Drug Dispos. 2013, 34, 29–44.
- Lamhonwah, A.M.; Ackerley, C.A.; Tilups, A.; Edwards, V.D.; Wanders, R.J.; Tein, I. OCTN3 is a mammalian peroxisomal membrane carnitine transporter. Biochem. Biophys. Res. Commun. 2005, 338, 1966–1972.
- Stanley, C.A.; DeLeeuw, S.; Coates, P.M.; Vianey-Liaud, C.; Divry, P.; Bonnefont, J.P.; Saudubray, J.M.; Haymond, M.; Trefz, F.K.; Breningstall, G.N.; et al. Chronic cardiomyopathy and weakness or acute coma in children with a defect in carnitine uptake. Ann. Neurol. 1991, 30, 709–716.
- Ingoglia, F.; Visigalli, R.; Rotoli, B.M.; Barilli, A.; Riccardi, B.; Puccini, P.; Dall’Asta, V. Functional activity of l-carnitine transporters in human airway epithelial cells. Biochim. Biophys. Acta 2016, 1858, 210–219.
- Tein, I.; Bukovac, S.W.; Xie, Z.W. Characterization of the human plasmalemmal carnitine transporter in cultured skin fibroblasts. Arch. Biochem. Biophys. 1996, 329, 145–155.
- Nakanishi, T.; Hatanaka, T.; Huang, W.; Prasad, P.D.; Leibach, F.H.; Ganapathy, M.E.; Ganapathy, V. Na+- and Cl- coupled active transport of carnitine by the amino acid transporter ATB(0,1) from mouse colon expressed in HRPE cells and Xenopus oocytes. J. Physiol. 2001, 532, 297–304.
- Enomoto, A.; Wempe, M.F.; Tsuchida, H.; Shin, H.J.; Cha, S.H.; Anzai, N.; Goto, A.; Sakamoto, A.; Niwa, T.; Kanai, Y.; et al. Molecular identification of a novel carnitine transporter specific to human testis. Insights into the mechanism of carnitine recognition. J. Biol. Chem. 2002, 277, 36262–36271.
- Georges, B.; Le Borgne, F.; Galland, S.; Isoir, M.; Ecosse, D.; Grand-Jean, F.; Demarquoy, J. Carnitine transport into muscular cells. Inhibition of transport and cell growth by mildronate. Biochem. Pharmacol. 2000, 59, 1357–1363.
- El-Hattab, A.W.; Scaglia, F. Disorders of carnitine biosynthesis and transport. Mol. Genet. Metab. 2015, 116, 107–112.
- Jain, S.S.; Luiken, J.J.; Snook, L.A.; Han, X.X.; Holloway, G.P.; Glatz, J.F.; Bonen, A. Fatty acid transport and transporters in muscle are critically regulated by Akt2. FEBS Lett. 2015, 589, 2769–2775.
- Radif, Y.; Ndiaye, H.; Kalantzi, V.; Jacobs, R.; Hall, A.; Minogue, S.; Waugh, M.G. The endogenous subcellular localisations of the long chain fatty acid-activatingenzymes ACSL3 and ACSL4 in sarcoma and breast cancer cells. Mol. Cell. Biochem. 2018, 448, 275–286.
- Jia, Z.; Pei, Z.; Maiguel, D.; Toomer, C.J.; Watkins, P.A. The fatty acid transport protein (FATP) family: Very long chain acyl-CoA synthetases or solute carriers? J. Mol. Neurosci. 2007, 33, 25–31.
- Hoppel, C.L. Carnitine and carnitine palmitoyltransferase in fatty acid oxidation and ketosis. Fed. Proc. 1982, 41, 2853–2857.
- Rufer, A.C.; Thoma, R.; Hennig, M. Structural insight into function and regulation of carnitine palmitoyltransferase. Cell. Mol. Life Sci. 2009, 66, 2489–2501.
- Serviddio, G.; Giudetti, A.M.; Bellanti, F.; Priore, P.; Rollo, T.; Tamborra, R.; Siculella, L.; Vendemiale, G.; Altomare, E.; Gnoni, G.V. Oxidation of hepatic carnitine palmitoyl transferase-I (CPT-I) impairs fatty acid beta-oxidation in rats fed a methionine-choline deficient diet. PLoS ONE 2011, 6, e24084.
- Woldegiorgis, G.; Shi, J.; Zhu, H.; Arvidson, D.N. Functional characterization of mammalian mitochondrial carnitine palmitoyltransferases I and II expressed in the yeast Pichia pastoris. J. Nutr. 2000, 130, 310S–314S.
- Lee, J.; Wolfgang, M.J. Metabolomic profiling reveals a role for CPT1c in neuronal oxidative metabolism. BMC Biochem. 2012, 13, 23.
- Foster, D.W. Malonyl-CoA: The regulator of fatty acid synthesis and oxidation. J. Clin. Investig. 2012, 122, 1958–1959.
- Zeth, K.; Thein, M. Porins in prokaryotes and eukaryotes: Common themes and variations. Biochem. J. 2010, 431, 13–22.
- Indiveri, C.; Iacobazzi, V.; Tonazzi, A.; Giangregorio, N.; Infantino, V.; Convertini, P.; Console, L.; Palmieri, F. The mitochondrial carnitine/acylcarnitine carrier: Function, structure and physiopathology. Mol. Asp. Med. 2011, 32, 223–233.
- Tonazzi, A.; Galluccio, M.; Oppedisano, F.; Indiveri, C. Functional reconstitution into liposomes and characterization of the carnitine transporter from rat liver microsomes. Biochim. Biophys. Acta 2006, 1758, 124–131.
- Giudetti, A.M.; Stanca, E.; Siculella, L.; Gnoni, G.V.; Damiano, F. Nutritional and hormonal regulation of citrate and carnitine/acylcarnitine transporters: Two mitochondrial carriers involved in fatty acid metabolism. Int. J. Mol. Sci. 2016, 25, 817.
- Priore, P.; Stanca, E.; Gnoni, G.V.; Siculella, L. Dietary fat types differently modulate the activity and expression of mitochondrial carnitine/acylcarnitine translocase in rat liver. Biochim. Biophys. Acta 2012, 1821, 1341–1349.
- Cavallo, A.; Priore, P.; Gnoni, G.V.; Papa, S.; Zanotti, F.; Gnoni, A. 3,5-Diiodo-L-thyronine administration to hypothyroid rats rapidly enhances fatty acid oxidation rate and bioenergetic parameters in liver cells. PLoS ONE 2013, 8, e52328.
- Cavallo, A.; Taurino, F.; Damiano, F.; Siculella, L.; Sardanelli, A.M.; Gnoni, A. Acute administration of 3,5-diiodo-L-thyronine to hypothyroid rats stimulates bioenergetic parameters in liver mitochondria. J. Bioenerg. Biomembr. 2016, 48, 521–529.
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197.
- Violante, S.; Ijlst, L.; Te Brinke, H.; Koster, J.; Tavares de Almeida, I.; Wanders, R.J.; Ventura, F.V.; Houten, S.M. Peroxisomes contribute to the acylcarnitine production when the carnitine shuttle is deficient. Biochim. Biophys. Acta 2013, 1831, 1467–1474.
- Schrader, M.; Costello, J.; Godinho, L.F.; Islinger, M. Peroxisome-mitochondria interplay and disease. J. Inherit. Metab. Dis. 2015, 38, 681–702.
- Schönfeld, P.; Wojtczak, L. Short- and medium-chain fatty acids in energy metabolism: The cellular perspective. J. Lipid Res. 2016, 57, 943–954.
This entry is offline, you can click here to edit this entry!