Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
HMGB1, originally described as a a protein that binds to DNA, functions as a structural co-factor for somatic cell transcription control. However, it also has numerous functions extracellularly. High mobility group box 1 (HMGB1), when passively released from cells, is capable of activating host innate immunity.
- HMGB1
- keratitis
- silencing HMGB1
- anti-HMGB1 antibody
- HMGB1 box A
- blockade of receptors
- thrombomodulin
- VIP
- glycyrrhizin
1. Introduction
Numerous reports have suggested that HMGB1 is a promising target for therapeutic intervention inanimal models of corneal infection using P. aeruginosa- [3][4], pneumonia in cystic fibrosis patients [1], in sepsis [5][6][7], arthritis [8], and other diseases [9]. HMGB1 is a ligand for receptor for advanced glycation end products (RAGE) and induces nuclear translocation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) in macrophages (Mϕ), and neutrophils (PMN). It is found readily in the sputum of cystic fibrosis patients [1] and in the serum of septic patients, where elevated levels are consistent with poor prognosis [5]. Monocytes [10], Mϕ, [10][11], natural killer cells [12] and dendritic cells [13] secrete HMGB1 in response to engaging pathogen associated molecular patterns (PAMPS) (e.g., lipopolysaccharide, LPS), potentiating immunity.
HMGB1 structure dictates how it functions (Figure 1). It is a 215 amino acid protein with a tendency to bind LPS, and cytokines such as interleukin (IL)-1 (α and β), DNA, histones, and other molecules. Its two major receptors are Toll-like receptor (TLR) 4 and RAGE. HMGB1 acts alone or in complex with the latter molecules [14]. If HMGB1 acts on its own, its redox state, which depends on its 3 cysteines is key for a proinflammatory response. In an unstimulated cell, HMGB1 in the nucleus is fully reduced (the three cysteines express thiol groups). HMGB1 released extracellularly, in the fully reduced form, complexes with the C-X-C motif chemokine (CXC) ligand (L) 12 to enhance cell chemotaxis by binding to the CXC receptor (R) 4 [3][14]. The HMGB1 isoform with a disulfide linkage between C 23 and C 45, at the same time that C 106 remains in its reduced form as a thiol (Figure 1) allows the molecule to activate proinflammatory cytokine production. This occurs by interacting with the Toll like receptor (TLR)4 receptor, through binding to myeloid differentiation factor 2 (MD-2) [14]. Disulfide HMGB1 loses its ability to activate TLR4 in either reduced or further oxidized; reduced and disulfide isoforms are reversible. Further oxidation of HB1 generates an irreversibly converted molecule with no proinflammatory capacity [14].
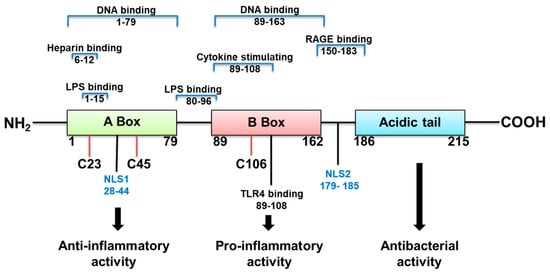
Figure 1. HMGB1 structure, binding sites and function.
HMGB1 released to the extracellular space is an attractive candidate for therapy, because of its function as a late mediator of inflammation, with levels highest levels attained at 2–3 days after infection [7]. Here, we review studies examining inhibition of HMGB1 as a therapeutic target in P. aeruginosa keratitis with some comparisons to other systems.
2. P. aeruginosa Keratitis: Role of Immune System in Disease
P. aeruginosa is a Gram-negative bacterium, often described as an opportunistic pathogen, and an important human pathogen as well. In developed countries, it remains the most common organism causing contact lens-related keratitis, one of the most rapidly developing and potentially blinding diseases of the cornea [15]. About 140 million people wear lenses worldwide, making P. aeruginosa-induced keratitis a global cause of visual impairment and blindness. In the USA alone, the cost of P. aeruginosa keratitis is approximately $175 million in direct healthcare expenditures, imposing both a clinical and an economic burden [15][16][17][18][19][20]. P. aeruginosa corneal infection induces inflammatory epithelial edema, stromal infiltration/destruction, ulceration and ultimately, vision loss [15].
Innate immunity exerts a major and critical component in the host pathogenic response to P. aeruginosa [15][21][22][23][24][25][26]. PMN and Mϕ are recruited to the infection site, engulf bacteria, and kill them by producing reactive oxygen (ROS) and nitrogen (RNS) species and facilitate their clearance [15][21][22][23][24][25]. PMN, the predominant infiltrating cell, are critical for microbial clearance [15][21][22][23][24][25]. Often, however, their persistence results in increased tissue damage and corneal perforation [21][22][23][24][25][26]. Mϕ curtail bacterial growth and regulate immune responses by controlling PMN infiltration, apoptosis and balancing pro- and anti-inflammatory cytokines and other cell responses [26][27]. This was shown by depleting mice of Mϕ by subconjunctival injection of clodronate-containing liposomes. These mice exhibited an increased influx of PMN into the cornea and more severe keratitis [27]. The adaptive immune system also is involved in this disease. It has been shown that a T helper 1 (Th1)-dominant response is associated with genetic susceptibility, severe corneal disease and perforation. In contrast, a T helper 2 (Th2)-dominant response leads to resistance, a milder course of disease and no perforation [28][29][30][31]. Recent evidence also has shown that Th17 cells infiltrate the cornea in the late stage of P. aeruginosa infection, sustaining inflammation, PMN influx and development of more severe disease [32][33].
3. P. aeruginosa Conventional Treatment
Antibiotic Treatment
Treatment of P. aeruginosa keratitis involves intensive topical antimicrobial therapy using fluoroquinolones (e.g., moxifloxacin) or fortified Gram-negative antibiotics, including aminoglycosides (e.g., tobramycin), cephalosporins (e.g., ceftazidime), and synthetic penicillins (e.g., carbenicillin) and in severe cases, by their subconjunctival injection. Although antibiotics reduce bacterial burden, tissue damage occurs due to a poorly controlled host immune response [34][35]. Additionally, antibiotic resistant bacteria are continuously emerging and pose serious challenges for the effective management of keratitis [36]. Resistance to antimicrobials has been noted from the time the first antibiotics were discovered, and many genes that confer drug resistance upon some strains of bacteria pre-date antibiotics by millions of years [37]. However, resistance has increasingly become problematic globally due to overuse of antimicrobials which has contributed to increasing the rate of resistance development and spread. The lack of new drugs to challenge these new “supermicrobes” exacerbates the problem. Besides treatment issues, there also is an economic impact of this growing problem, as more than 2 million infections a year are caused by bacteria that are resistant to first-line antibiotics, [37] costing the US health system 20 billion dollars each year [38]. P. aeruginosa, an opportunistic pathogen, that does not normally infect healthy individuals, but rather those who are hospitalized or immunocompromised in some way, causes 51,000 infections/year in the USA; 13 percent [37] of these are multi-drug resistant (MDR) and increasingly difficult to treat. Other approaches, such as inhibition of HMGB1, provide novel alternatives to prevent and treat infections resulting from both resistant and non-resistant bacteria; it is both pressing and timely that we develop these alternative treatments.
This entry is adapted from the peer-reviewed paper 10.3390/pathogens10101235
References
- Entezar, M.; Weiss, D.J.; Sitapara, R.; Whittaker, L.; Wargo, M.J.; Li, J.; Wang, H.; Yang, H.; Sharma, L.; Phan, B.D.; et al. Inhibition of high-mobility group box 1 protein (HMGB1) enhances bacterial clearance and protects against Pseudomonas aeruginosa pneumonia in cystic fibrosis. Mol. Med. 2012, 18, 477–485.
- Reeves, R. Nuclear functions of the HMG proteins. Biochim. Biophys. Acta. 2010, 1799, 3–14.
- McClellan, S.; Jiang, X.; Barrett, R.; Hazlett, L.D. High-mobility group box 1: A novel target for treatment of Pseudomonas aeruginosa keratitis. J. Immunol. 2015, 194, 1776–1787.
- Ekanayaka, S.A.; McClellan, S.A.; Barrett, R.P.; Kharotia, S.; Hazlett, L.D. Glycyrrhizin reduces HMGB1 and bacterial load in Pseudomonas aeruginosa keratitis. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5799–5809.
- Huang, W.; Tang Li, L. HMGB1, a potent proinflammatory cytokine in sepsis. Cytokine 2010, 51, 119–126.
- Czura, C.; Yang, H.; Amella, C.A.; Tracey, K.J. HMGB1 in the immunology of sepsis (Not septic shock) and arthritis. Adv. Immunol. 2004, 84, 181–200.
- Wang, H.; Bloom, O.; Zhang, M.; Vishnubhakat, J.M.; Ombrellino, M.; Che, J.; Frazier, A.; Yang, H.; Ivanova, S.; Borovikova, L.; et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999, 285, 248–251.
- Andersson, E. Landsson-Harris H. HMGB1 is a potent trigger of arthritis. J. Intern. Med. 2004, 255, 344–350.
- Zhang, F.; Huang, G.; Hu, B.; Fang, L.P.; Cao, E.; Xin, X.F.; Song, Y.; Shi, Y. Anti-HMGB1 neutralizing antibody ameliorates neutrophilic airway inflammation by suppressing dendritic cell-mediated Th17 polarization. Mediat. Inflamm. 2014, 2014.
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195.
- Tang, D.; Shi, Y.; Kang, R.; Li, T.; Xiao, W.; Wang, H.; Xiao, X. Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J. Leuko. Biol. 2007, 81, 741–747.
- Li, G.; Liang, X.; Lotze, M.T. HMGB1: The central cytokine for all lymphoid cells. Front. Immunol. 2013, 4, 68.
- Dumitriu, I.E.; Baruah, P.; Valentinis, B.; Voll, R.E.; Herrmann, M.; Nawroth, P.P.; Arnold, B.; Bianchi, M.E.; Manfredi, A.A.; Rovere-Querini, P. Release of high mobility group box 1 by dndritic cells controls T cell activation via the receptor for advanced glycation end products. J. Immunol. 2005, 174, 7506–7515.
- Yang, H.; Wang, H.; Andersson, U. Targeting inflammation driven by HMGB1. Front. Immunol. 2020, 11, 1–9.
- Hazlett, L.D. Corneal response to Pseudomonas aeruginosa infection. Prog. Retin. Eye Res. 2004, 23, 1–30.
- Choy, M.H.; Stapleton, F.; Willcox, M.D.; Zhu, H. Comparison of virulence factors in Pseudomonas aeruginosa strains isolated from contact lens-and non-contact lens-related keratitis. J. Med. Microbiol. 2008, 57, 1539–1546.
- Stapleton, F.; Carnt, N. Contact lens-related microbial keratitis: How have epidemiology and genetics helped us with pathogenesis and prophylaxis. Eye 2012, 26, 185–193.
- Green, M.; Apel, A.; Stapleton, F. Risk factors and causative organisms in microbial keratitis. Cornea 2008, 27, 22–27.
- Tam, C.; Mun, J.J.; Evans, D.J.; Fleiszig, S.M. The impact of inoculation parameters on the pathogenesis of contact lens-related infectious keratitis. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3100–3106.
- Ung, L.; Bispo, P.J.M.; Shanbhag, S.S.; Gilmore, M.S.; Chodosh, J. The persistent dilemma of microbial keratitis: Global burden, diagnosis and antimicrobial resistance. Surv. Ophthalmol. 2019, 64, 255–271.
- Kernacki, K.A.; Barrett, R.P.; Hobden, J.A.; Hazlett, L.D. Macrophage inflammatory protein-2 is a mediator of polymorphonuclear neutrophil influx in ocular bacterial infection. J. Immunol. 2000, 164, 1037–1045.
- Kernacki, K.A.; Barrett, R.P.; McClellan, S.; Hazlett, L.D. MIP-1alpha regulates CD4+ T cell chemotaxis and indirectly enhances PMN persistence in Pseudomonas aeruginosa corneal infection. J. Leukoc. Biol. 2001, 70, 911–919.
- Hazlett, L.D. Pathogenic mechanisms of P. aeruginosa keratitis: A review of the role of T cells, Langerhans cells, PMN, and cytokines. DNA Cell Biol. 2002, 21, 383–390.
- Rudner, X.L.; Kernacki, K.A.; Barrett, R.P.; Hazlett, L.D. Prolonged elevation of IL-1 in Pseudomonas aeruginosa ocular infection regulates macrophage-inflammatory protein-2 production, polymorphonuclear neutrophil persistence, and corneal perforation. J. Immunol. 2000, 164, 6576–6582.
- Kernacki, K.A.; Barrett, R.P.; McClellan, S.A.; Hazlett, L.D. Aging and PMN response to P. aeruginosa infection. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3019–3025.
- Zhou, Z.; Barrett, R.P.; McClellan, S.A.; Zhang, Y.; Szliter, E.A.; van Rooijen, N.; Hazlett, L.D. Substance P delays apoptosis, enhancing keratitis after Pseudomonas aeruginosa infection. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4458–4467.
- McClellan, S.A.; Huang, X.; Barrett, R.P.; van Rooijen, N.; Hazlett, L.D. Macrophages restrict Pseudomonas aeruginosa growth, regulate polymorphonuclear neutrophil influx, and balance pro- and anti-inflammatory cytokines in BALB/c mice. J. Immunol. 2003, 170, 5219–5227.
- Kwon, B.; Hazlett, L.D. Association of CD4+ T cell-dependent keratitis with genetic susceptibility to Pseudomonas aeruginosa ocular infection. J. Immunol. 1997, 159, 6283–6290.
- Hazlett, L.; McClellan, S.; Kwon, B.; Barrett, R. Increased severity of Pseudomonas aeruginosa corneal infection in strains of mice designated as Th1 versus Th2 responsive. Investig. Ophthalmol. Vis. Sci. 2000, 41, 805–810.
- Berk, R.S.; Leon, M.A.; Hazlett, L.D. Genetic control of the murine corneal response to Pseudomonas aeruginosa. Infect. Immun. 1979, 26, 1221–1223.
- Berk, R.S.; Beisel, K.; Hazlett, L.D. Genetic studies of the murine corneal response to Pseudomonas aeruginosa. Infect. Immunity 1981, 34, 1–5.
- Suryawanshi, A.; Cao, Z.; Thitiprasert, T.; Zaidi, T.S.; Panjwani, N. Galectin-1-mediated suppression of Pseudomonas aeruginosa—Induced corneal immunopathology. J. Immunol. 2013, 190, 6397–6409.
- Li, C.; McClellan, S.A.; Barrett, R.; Hazlett, L.D. Interleukin 17 regulates Mer tyrosine kinase-positive cells in Pseudomonas aeruginosa keratitis. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6886–6900.
- Gadjeva, M.; Nagashima, J.; Zaidi, T.; Mitchell, R.A.; Pier, G.B. Inhibition of macrophage migration inhibitory factor ameliorates ocular Pseudomonas aeruginosa—Induced keratitis. PLoS Pathog. 2010, 6, e1000826.
- O’Brien, T.P.; Maguire, M.G.; Fink, N.E.; Alfonso, E.; McDonnell, P. Efficacy of ofloxacin vs. cefazolin and tobramycin in the therapy for bacterial keratitis. Report from the Bacterial Keratitis Study Research Group. Arch. Ophthalmol. 1995, 113, 1257–1265.
- Mesaros, N.; Nordmann, P.; Plesiat, P.; Roussel-Delvallez, M.; Van Eldere, J.; Glupczynski, Y.; Van Laethem, Y.; Jacobs, F.; Lebecque, P.; Malfroot, A.; et al. Pseudomonas aeruginosa: Resistance and therapeutic options at the turn of the new millennium. Clin. Microbiol. Infec. 2007, 13, 560–578.
- Centers for Disease Control and Prevention. Antibiotic resistance threats in the United States; CDC: Atlanta, GA, USA, 2013; pp. 1–114.
- Smith, R.; Coast, J. The true cost of antimicrobial resistance. BMJ 2013, 346, f1493.
This entry is offline, you can click here to edit this entry!