Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biology
Mitochondria play a key role in maintaining energy homeostasis in metabolic tissues, including adipose tissues. The two main types of adipose tissues are the white adipose tissue (WAT) and the brown adipose tissue (BAT). WAT primarily stores excess energy, whereas BAT is predominantly responsible for energy expenditure by non-shivering thermogenesis through the mitochondria. WAT in response to appropriate stimuli such as cold exposure and β-adrenergic agonist undergoes browning wherein it acts as BAT, which is characterized by the presence of a higher number of mitochondria.
- mitochondria
- mitochondrial dysfunction
1. Introduction
Adipose tissue is the major organ that controls the overall energy homeostasis in a living organism. Briefly, in excess energy conditions, the adipose tissue stores the superabundant nutrients in the form of triglycerides, whereas during scarcity of energy, it supplies the nutrients to other tissues through lipolysis [13]. In mammals, there are two different types of adipose tissues, namely, white adipose tissue (WAT) and brown adipose tissue (BAT). Interestingly, WAT and BAT have opposite functions. The WAT stores excess energy as triglycerides, while the BAT is specialized in the dissipation of energy through heat production for maintaining the body temperature and energy consumption. Recent studies have demonstrated that beige adipocytes sporadically reside with white adipocytes and emerge in response to certain environmental cues. Beige adipocytes have the antagonistic functions of WAT and agonistic functions of BAT at the same time. In the basal state, beige adipocytes act as white adipocytes; however, when given signals that require heat production through energy consumption such as cold stimulation, these cells demonstrate BAT-like function and morphology [14].
2. Role of the Mitochondria in Adipocytes
2.1. Adipocyte Differentiation
Mitochondria are key organelles that control the physiological role of adipocytes such as adipocyte differentiation, lipid homeostasis, insulin sensitivity, oxidative capacity, adaptive thermogenesis, and browning of WATs (Figure 1). In particular, adipocyte differentiation is characterized by an induction in mitochondrial metabolism. During adipogenesis, preadipocytes undergo sequential transcriptional regulation by adipogenic regulators such as peroxisome proliferator-activated receptor-γ (PPARγ), CCAAT-enhancer-binding proteins (C/EBPs), and PPARγ coactivator 1-α (PGC1α), thereby leading to the promotion of mitochondrial biogenesis [35]. In addition, mitochondrial remodeling is involved in the quantitative and qualitative changes of the mitochondria during adipocyte differentiation [36]. Within 4 days of adipogenic induction of 3T3-L1, which are immortalized white preadipocytes, the expression of mitochondrial proteins and the number of mitochondria increases dramatically and is accompanied by qualitative changes in the mitochondrial composition, including pyruvate carboxylase and aconitase, which are involved in fatty acid metabolism [37]. Moreover, the increased oxygen consumption rate of the differentiated adipocytes compared to that of the preadipocytes is clear evidence that mitochondria are activated during adipocyte differentiation [38].
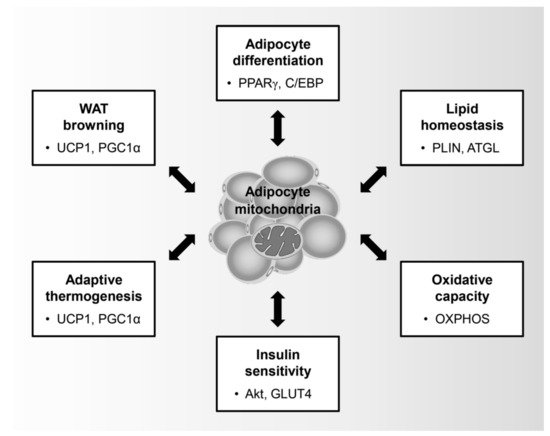
Figure 1. Physiological role of adipocyte mitochondria: Mitochondria in adipocytes regulate adipocyte differentiation, lipid homeostasis, oxidative capacity, insulin sensitivity, adaptive thermogenesis, and browning of white adipose tissues. Peroxisome proliferator-activated receptor-γ (PPARγ); CCAAT-enhancer-binding protein (C/EBP); Perilipin (PLIN); Adipose triglyceride lipase (ATGL); Oxidative phosphorylation (OXPHOS); Glucose transporter type 4 (GLUT4); Uncoupling protein-1 (UCP1); PPARγ coactivator 1-α (PGC1α).
Previous studies have shown that the induction of mitochondria is essential for ATP production, which is necessary for increasing the metabolic activity during the adipogenic program. Despite promoted mitochondrial activity during adipocyte differentiation, the ATP content is decreased by high ATP consumption processes such as lipogenesis [37]. In the adipogenic program, the TCA cycle produces reducing equivalents such as nicotinamide adenine dinucleotide and flavin adenine dinucleotide and achieves oxidation of the acetyl-coenzyme A—an important metabolite produced from the catabolism of glucose and fatty acids [21]. Recently, it was demonstrated that mitochondrial dysfunction adversely affects adipocyte differentiation. Specific inhibitors of mitochondrial di- and tricarboxylate carriers prevent the differentiation of 3T3-L1 adipocytes, suggesting that mitochondria play an essential role in adipogenesis [39].
Mitochondrial reactive oxygen species (ROS) produced by the respiratory chain act as a second messenger and plays a crucial role in numerous cellular signaling pathways inside and outside the mitochondria. Mitochondrial ROS produced by OXPHOS complex III is essential for the initiation of adipocyte differentiation of human mesenchymal stem cells, whereas adipogenesis is inhibited by mitochondria-targeted antioxidants [23]. However, high doses of ROS negatively regulate the proliferation and differentiation of adipocytes [40]. In particular, increased ROS production by rotenone, a complex I inhibitor, or oligomycin, a F0-F1 ATP synthase inhibitor, has been reported to prevent the adipocyte differentiation of human mesenchymal stem cells [41,42]. A number of studies support the importance of mitochondria in adipocyte differentiation. Therefore, it is rational to target mitochondria for the treatment of obesity caused by the abnormal control of adipocyte differentiation.
2.2. Lipid Homeostasis and Oxidative Capacity
Adipose tissues play a key role in maintaining whole-body lipid homeostasis. Adipose tissues store lipids in the form of triglycerides in lipid droplets, thereby preventing ectopic lipid accumulation in other organs. Perilipin (PLIN), a lipid droplet coat protein, is required for the stable storage of lipid metabolites in adipocytes [43]. The triglycerides are broken into fatty acids and glycerol by adipose triglyceride lipase (ATGL)-mediated lipolysis, sequentially used as energy [44]. In obesity, excessive lipogenesis increases the total lipid pool, leading to insulin resistance and hyperglycemia [45]. It has been reported that the mitochondrial dysfunction with an OXPHOS complex III inhibitor causes triglyceride accumulation by inducing excessive lipogenesis in 3T3-L1 preadipocytes [46]. In particular, mitochondria in brown adipocytes are involved in lipid homeostasis by controlling fatty acid oxidation, which is one of the key pathways for lipid clearance [47]. Therefore, ectopic lipid accumulation is promoted by reducing fatty acid oxidation and energy expenditure due to mitochondrial dysfunction in the brown adipocytes of people with obesity and metabolic diseases [48].
A previous study showed that the abundance of mitochondrial populations is halved in adipocytes isolated from epididymal adipose tissues from obese and diabetic ob/ob mice [49]. Moreover, two obese and diabetic mouse models—the genetic model db/db mice and the dietary high-fat diet-fed mice—showed a decreased level of mitochondrial biogenesis regulators, including PGC1α and estrogen-related receptor-α, resulting in the loss of mitochondrial mass and structure [50]. Furthermore, mitochondrial dysfunction caused by tumor necrosis factor-α (TNF-α), which is an inflammatory cytokine that increases in obesity, leads to smaller and condensed mitochondria and inhibits intracellular ATP synthesis in 3T3-L1 adipocytes [51]. In addition, the expression of genes involved in OXPHOS complexes and fatty acid oxidation is decreased by TNF-α in primary adipocytes [52]. Under various pathological conditions, the oxidative capacity, biogenesis, density, and dynamics of mitochondria could be disrupted in the adipocytes, resulting in the development of obesity and metabolic diseases.
The main function of mitochondria in the adipocytes is to produce ATP to support a variety of metabolic pathways, including triglyceride synthesis, gluconeogenesis, and fatty acid re-esterification [21]. Dysregulation of mitochondria destroys the oxidative capacity and eventually fails to generate ATP. The mitochondrial membrane potential and the activity of the respiratory chain complexes are reported to be significantly decreased in the subcutaneous adipose tissues of obese subjects [53]. Moreover, the expression of the genes involved in mitochondrial oxidative pathways, including fatty acid oxidation, TCA cycle, ketogenesis, ketolysis, and branched chain amino acid degradation, is suppressed in obese individuals. One study showed that the protein levels of OXPHOS complexes III, IV, and V are also decreased in the adipose tissues of obese subjects [54]. Inverse correlations have been reported between body mass index and mitochondrial respiratory capacity in human adipose tissues. Furthermore, ADP-stimulated mitochondrial respiration, mtDNA copy number, and OXPHOS complex protein expression are dramatically decreased in the adipose tissues of obese subjects [55]. Thus, previous studies have shown the correlation between mitochondrial oxidative capacity in the adipocytes and systemic energy metabolism.
2.3. Glucose Utilization and Insulin Sensitivity
Insulin is a major anabolic hormone also involved in the regulation of energy and lipid metabolism through the phosphatidylinositol 3-kinase (PI3K)–Akt signaling pathway. The primary function of insulin is to allow glucose uptake into muscle cells and adipocytes to lower the blood glucose level. Binding of insulin to its receptor on adipocytes triggers the phosphorylation and activation of insulin receptor substrate (IRS), and it forms a docking site for PI3K at the cell membrane. When docked, PI3K converts phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3). Subsequently PIP3 activates phosphoinositide-dependent protein kinase 1 (PDK1) and Akt. Akt stimulates translocation of glucose transporter type 4 (GLUT4) translocation to cell membranes, thus allowing glucose to enter the cell [56]. Adipocyte-specific Glut4 knockout mice exhibit systemic glucose intolerance and insulin resistance [57].
Recently, mitochondria in adipocytes have been suggested to play a role in regulating insulin sensitivity. Treatments with respiratory inhibitors or uncoupling reagents that block mitochondrial function were shown to reduce insulin-stimulated glucose uptake in adipocytes [58]. In high-fat-diet-fed rats, mitochondrial biogenesis and the copy number of mitochondrial DNA (mtDNA) were decreased in epididymal adipose tissues, accompanied by hyperglycemia [59]. Further, high levels of glucose and/or free fatty acid attenuated insulin-stimulated glucose uptake by the dysfunctional mitochondria in the adipocytes. This glucose and/or free fatty acid-mediated reduction of glucose uptake was recovered by PGC1α overexpression, which is involved in the normalization of mitochondria [60]. The CR6-interacting factor-1 (Crif1) is a key translational factor responsible for the protein expression of mitochondrial OXPHOS complexes. Crif1 ablation in adipocytes causes loss of OXPHOS complex subunits and respiratory complexes, sequentially resulting in whole-body insulin resistance [61]. These studies suggest that mitochondria in adipocytes are required for regulating glucose use through insulin signaling pathway.
2.4. Thermogenesis and WAT Browning
Recent studies have focused on the role of mitochondria in brown adipocytes that promotes energy consumption via adaptive thermogenesis [62]. Activation of mitochondria in brown adipocytes accelerates heat generation by increasing the inner mitochondrial membrane UCP1 [30]. Activated UCP1 uncouples the electron transport of the respiratory chain, thereby blocking ATP production and dissipating energy in the form of heat [63]. It has been demonstrated that the activity of UCP1 and thermogenesis in mouse BAT is associated with body-weight control and energy homeostasis [64]. Moreover, the adipocyte UCP1 expression is reduced in obese subjects, while metabolic complications are improved with UCP1 activation by various environmental and pharmacological stimuli [65]. Strategies for combating metabolic diseases through the activation of UCP1 have been proposed; for instance, eight subjects with diabetes reported a 43% increase in insulin sensitivity when UCP1 was activated by cold acclimation (14–15 °C) for 10 days [66]. Thus, adaptive thermogenesis-dependent fat burn has emerged as a plausible and safe strategy for relieving the metabolic syndrome.
The most important and well-studied factor that activates thermogenesis is norepinephrine (NE). NE affects cell proliferation and differentiation as well as thermogenesis of brown adipocytes. NE increases cyclic adenosine monophosphate levels via the β3-adrenergic receptor and activates protein kinase A, subsequently leading to lipolysis-mediated production of free fatty acids, one of the acute substrates of thermogenesis [67]. PGC1α has been reported to be essential for thermogenic activation by cold-induced and β3-adrenergic agonists in brown adipocytes [68]. PGC1α was first identified as a cold-induced interacting partner of PPARγ in BAT [69]. Mechanistically, PGC1α promotes mitochondrial biogenesis and oxidative metabolism by inducing UCP1 transcription [67]. Notably, thermogenic activation in brown adipocytes is accompanied by mitochondrial fission and depolarization [70]. These studies suggest that thermogenesis might be regulated by mitochondrial dynamics in brown adipocytes.
A recent study showed that white adipocytes, which have low UCP1 expression and low mitochondrial contents, sometimes act as inducible thermogenic adipocytes that increase UCP1 expression and energy consumption under certain conditions such as cold exposure and β-adrenergic activation. Beige adipocytes (also called “brite”, “brown-like”, or “inducible brown”) are generated by the browning of WAT [71] (Figure 2). Beige adipocytes exhibit UCP1-dependent thermogenesis, and these mitochondria use lipids or carbohydrates as substrates, similar to brown adipocytes [72]. Chronic exercise decreases body weight and fat mass by inducing browning of subcutaneous WAT [73]. In addition, Irisin, a PGC1α-dependent myokine induced by exercise, has been shown to increase energy expenditure and to improve insulin sensitivity by activation of WAT browning [74]. In adults with low compositions of BAT, increased energy expenditure through WAT browning is an attractive strategy for preventing metabolic diseases. Taken together, mitochondria are essential organelles for maintaining the function of adipocytes in metabolic homeostasis.
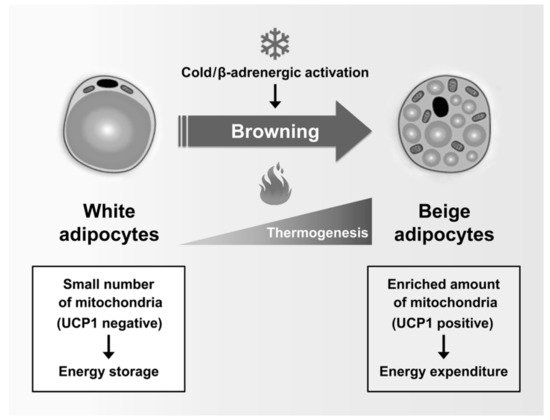
Figure 2. Regulation of adipose tissue browning: Beige adipocytes are generated by the browning of white adipose tissues in response to numerous stimuli, including cold exposure and activation of β-adrenergic receptors. Subsequently, the transcriptional machinery of the browning program activates the expression of characteristic thermogenic genes, leading to the formation of beige adipocytes. Similar to brown adipocytes, beige adipocytes are rich in mitochondria that express UCP1 and can achieve thermogenesis. Notably, beige adipocytes primarily contribute to energy expenditure rather than to energy storage.
This entry is adapted from the peer-reviewed paper 10.3390/ijms20194924
This entry is offline, you can click here to edit this entry!