3.1. Overview
A study showed that the activity of CaCCs, in general, was increased in the nasal epithelium in vivo of patients with CF [
71]. In 1986, another team demonstrated the presence of chloride current at the apical membrane of epithelial cells of CF in the presence of ionized calcium [
72]. Moreover, another study showed a decrease in ATP-induced chloride efflux in the primary bronchial epithelial cell line [
73]. The identification of ANO1 as a CaCC later on and its involvement in many deregulated processes in patients with CF made it a real therapeutic target. In the last few years, more research has been dedicated to ANO1 in CF.
Recent data reported the absence of calcium-induced chloride currents in epithelial cells of ANO1 KO mice [
74]. A subsequent study showed that CFTR expression requires ANO1 in plasma membranes, indicating a close relationship between the two chloride channels [
75]. Moreover, ANO1 expression and chloride activity were decreased in CF [
63]. According to several observations ((1) ANO1’s absence decreases airway secretion, (2) ANO1 is ubiquitously expressed in all the tissue affected by CF, including airway epithelial cells where ANO1 is a secondary chloride channel, (3) ANO1 provides a chloride pathway that is CFTR-independent, and (4) ANO1 is involved in HCO
3− secretion, which is highly important for fluid secretion and mucus hydration [
76]), increasing ANO1 activity can probably compensate for CFTR deficiency (
Figure 3). Different approaches targeting ANO1 have been developed to bypass CFTR dysfunction.
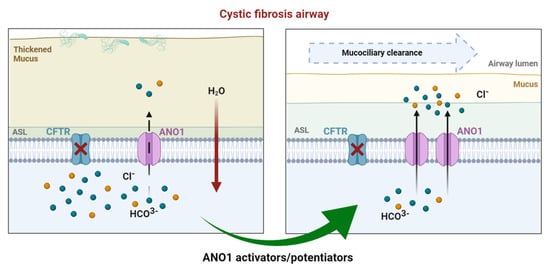
Figure 3. In CF airways, dysfunctional CFTR leads to compromised chloride efflux. Sodium entry is upregulated, leading to a dehydrated air surface liquid (ASL) and impaired mucociliary clearance favoring mucostasis, causing chronic inflammation and infection. In healthy airways, ANO1 is colocalized with CFTR within the apical membrane of epithelial cells, contributing to ion and water homeostasis. In CF ciliated cells, the expression of ANO1 is also diminished. Modulating ANO1, as an alternative CF therapy, could compensate for defective CFTR and, thus, enhance fluid secretion by ciliated epithelial cells, regulating ASL height and pH.
3.2. Drug Approaches Targeting ANO1 in Cystic Fibrosis
Many molecules have been used to modulate ANO1’s activity, but few were shown to be efficient and, most importantly, specific to ANO1 (Table 1).
Table 1. Summary of ANO1 inhibitors and activators used in CF.
|
Inhibitors
|
Specificity
|
Assay
|
References
|
|
ANI9
|
Not specific
|
In vitro
|
[82,85]
|
|
CCinh-A01
|
Not specific
|
In vitro, in vivo
|
[86]
|
|
DIDS
|
Not specific
|
In vitro
|
[87]
|
|
Diphenylamine-2-carboxylate (DPC), 5-nitro-2-(3-phenylpropylamino) benzoic acid
|
Not specific
|
In vitro
|
[88]
|
|
Flufenamic acid
|
Not specific
|
In vitro
|
[89,90]
|
|
Niclosamide
|
Not specific
|
In vitro, in vivo
|
[91]
|
|
Niflumic acid
|
Not specific
|
In vitro, in vivo
|
[92]
|
|
Plumbagin
|
Not specific
|
In vitro
|
[93]
|
|
Quercetin
|
Not specific
|
In vitro, in vivo, clinical trial (phase II)
|
[94,95,96]
|
|
Tannic acid
|
Not specific
|
In vitro
|
[97]
|
|
T16ainh-A01
|
Specific
|
In vitro
|
[86]
|
|
Activators
|
|
|
|
|
Denufosol (INS37217)
|
Not specific
|
In vitro, in vivo, clinical trial (phase III failed)
|
[77,78,79,80,81,98,99]
|
|
Duramycine (MOLI1901)
|
Not specific
|
In vitro, in vivo, clinical trial (phase II failed)
|
[100]
|
|
Eact
|
Not specific
|
In vitro
|
[86]
|
|
ETD002 (or ETX001)
|
Specific
|
In vitro, in vivo, clinical trial (phase I)
|
[101]
|
|
Interleukin 4
|
Not specific
|
In vitro
|
[14]
|
|
Resveratrol
|
Not specific
|
In vitro, in vivo, clinical trial
|
[102,103,104]
|
|
TSB ANO1
|
Specific
|
In vitro, in vivo, preclinical
|
[83]
|
Long before discovering ANO1’s function in the airways, a clinical trial targeting CaCCs, in general, took place. Inspire Pharmaceuticals developed denufosol (INS37217), the first CFTR-independent drug for CF lung therapy, carried into clinical trials in 2001 [
77,
78,
79,
80]. The inhaled P2Y2 receptor agonist activated P2Y receptors and led to intracellular calcium activation of chloride efflux through CACCs. This increased airway epithelial chloride efflux in vitro, increasing airway fluid volume. However, the second phase III clinical trial of denufosol was disappointing as it did not achieve statistical significance for its primary efficacy endpoint in improving forced expiratory volume in 1 s (FEV1). This failure was due to (1) denufosol targeting all CaCCs, (2) its short half-life in vivo due to rapid degradation by ectonucleotidases, and (3) increased intracellular calcium stimulating goblet cell exocytosis that might have led to an increase in mucus in the airways. Therefore, CaCC activators need to target CaCCs directly without elevating cytoplasmic calcium for more targeted therapy and efficacy [
81].
ANO1 identification paved the way for the development of specific activator molecules which would, without modifying the calcium signaling, obtain a more sustained activation over time, leading to better efficiency.
Another novel drug is an ANO1 potentiator (ETD002) developed by Enterprise Therapeutics (based in the University of Sussex Innovation Center, UK) and acquired by Roche (Genentech) in October 2020. This inhaled molecule demonstrated an upregulation of ANO1, which boosts epithelial fluid secretion and mucus clearance in primary CF bronchial epithelial cells and ovine models. Unlike denufosol, intracellular calcium measurements checked that ETD002 did not affect calcium mobilization, coherent with a direct effect on ANO1. A phase I study to test the safety of ETD002 in healthy participants is in progress [
82]. The mechanism via which ETD002 potentiates ANO1 activity is still unclear.
Our team has also worked on an innovative alternative approach using a locked nucleic acid (LNA)-enhanced antisense oligonucleotide (ASO). A previous study demonstrated that microRNA (miR-9) contributes to the downregulation of ANO1 expression and activity by directly targeting its 3’UTR. ASO ANO1 binds to the 3’UTR target site of ANO1 mRNA, preventing miR-9 from gaining access to that site [
83]. ASO ANO1 has increased ANO1 expression, chloride activity, and mucus clearance in primary human CF cells and CF mice. Recent studies have suggested that ANO1 plays an essential role in mucus production [
84]. To date, we have not observed any significant increase in mucin 5AC (MUC5AC, the main component of respiratory mucus produced by goblet cells) or mucin 5B (MUC5B, gel-forming mucin that plays a key role in mucociliary clearance) expression in vitro.
Currently, ANO1 activation as a therapeutic target is subject to controversial opinions. Centeio et al. found an upregulation of ANO1 expression in submucosal glands, airway smooth muscles, and pulmonary blood vessels in CF and asthmatic inflamed lungs [
105]. Moreover, activating ANO1 with Eact, which also activates other channels, induced mucus production in airway goblet cells and bronchoconstriction in ovalbumin-sensitized asthmatic mice [
85], whereby activating ANO1 could worsen the pathology in inflammatory airway diseases. Instead, the authors demonstrated that ANO1 inhibition by niclosamide significantly reduced goblet cell metaplasia and mucus production in asthmatic mice, as well as inhibited MUC5AC expression in Calu-3 human submucosal cells, suggesting that ANO1 inhibition might be beneficial in inflammatory airway diseases [
105]. It is also important to note that a transcriptome meta-analysis revealed that CF and asthma pathways are highly divergent [
106]. Furthermore, the same group reported a defect of mucus secretion and accumulation in secretory cells in 2018 when ANO1 was knocked out in ciliated airway epithelial cells and intestinal goblet cells, highlighting the vital role of ANO1 in mucus secretion [
84]. Another study showed that ANO1 inhibitors reduced both mucus secretion and airway hyperactivity [
69].
On the other hand, using a human respiratory basal cell line (BCi-NS1.1), Simões et al. showed that MUC5AC production does not require ANO1, and their simultaneous upregulation is only circumstantial under cell proliferation [
107]. The authors also showed a decrease in ASL when inhibiting ANO1. Furthermore, while replying to Olschewski et al.’s concerns on increasing ANO1 activity, Danahay et al. declared that positive modulation of ANO1 induces no bronchospasm in the conscious sheep model nor affects vascular smooth muscle contraction (unpublished observations) [
108,
109]. Overall, ANO1’s possible role in mucus production remains obscure and evokes controversial opinions over the beneficial or deleterious results of stimulating the channel in CF.