TRIMbody-Away technique is a novel technology that could be utilized to acute and rapid destruction of specific intracellular proteins. This technology is based on a new type of fusion protein, designated as TRIMbody, by fusing the truncated form of tripartite motif 21 (TRIM21) with the nanobody. The truncated TRIM21 retained only the N-terminal RBCC domain and deleted the C-terminal PRY-SPRY domain. Therefore, TRIMbody possesses the functions of TRIM21 and mAbs, but has much smaller size. TRIMbody-Away technique could expand the landscape of the applications of degrader technologies and provide an alternative approach for potential therapeutic benefit in future.
1. Introduction
Protein depletion or degradation technologies are widely used for researchers to understand the biological functions of intracellular proteins, which could be achieved by either interfering with protein synthesis or inducing protein degradation for controlling intracellular protein levels [
1]. Traditional ways to disturb protein synthesis include manipulations of gene sequences by CRISPR/Cas9 genome editing technology, targeting mRNA transcripts by RNA interference (RNAi), and morpholino antisense oligonucleotides [
2,
3,
4,
5]. However, protein depletion by those approaches is indirect and depends on the inherent turnover of the protein, which may be time-consuming or result in depletion resistance for some long-lived proteins. To induce direct degradation of a protein of interest, a number of approaches have been designed harnessing the power and specificity of the intracellular protein degradation machinery, such as proteolysis-targeting chimaeras (PROTACs), lysosome-targeting chimaeras (LYTACs), dTAGs, chaperone-mediated autophagy targeting, and non-genetic IAP-dependent protein erasers (SNIPERs) [
6,
7,
8,
9,
10]. All of these approaches have limitations, and their successful application depends on the protein of target and the experimental model of choice.
Recently, a technology termed TRIM-Away has been developed to acutely and rapidly degrade endogenous proteins in mammalian cells without change of the genome or mRNA expression level, using anti-target antibodies and TRIM21, which belongs to the TRIM (tripartite motif-containing) family [
11,
12]. The large majority of TRIM family proteins contain an N-terminal RBCC motif [
13,
14,
15], followed by C-terminal domains of various length and diverse composition that are often used to target specific substrates and mediate diverse functions [
16,
17]. The structural arrangement of the RBCC motif is highly conserved within the TRIM protein family, while the C-terminal region is highly variable [
18,
19], of which the most common is the PRY-SPRY domain, also known as the B30.2 domain. TRIM21 has a mass of 54 kDa and consists of an N-terminal RBCC motif and a C-terminal PRY-SPRY domain [
20,
21]. The RBCC motif includes a RING domain with E3 ubiquitin ligase activity, a B-box domain, and a coiled-coil dimerization domain [
22,
23]. The PRY-SPRY domain binds to the Fc fragment of immunoglobulin with high affinity [
24,
25,
26]. Therefore, TRIM-Away technology mainly relies on antibodies entering cells, where they recognize the target protein and bind to TRIM21, leading to substrate ubiquitination and degradation [
27]. However, the conventional antibodies had relatively large size (150 kDa), which results in correspondingly low tissue penetration and inaccessibility of some sterically hindered epitopes [
28,
29], and limits the degradation efficacy of target protein in cells.
An attractive alternative is using smaller antibody fragments to replace full-size mAbs. Single-domain antibodies (sdAbs), also designated as VHHs or nanobodies, have a small size with only 15 kDa, resulting in unique advantages compared to mAbs, including larger number of accessible epitopes, relatively lower production costs, and improved biophysical properties [
30,
31]. Thus, we constructed a new type of fusion protein, designated as TRIMbody, by fusing the truncated form of TRIM21 with the nanobody. The truncated TRIM21 retained only the N-terminal RBCC domain and deleted the C-terminal PRY-SPRY domain. Therefore, TRIMbody possesses the functions of TRIM21 and mAbs, but has relatively small size.
2. Plasmids
HLTV-hTRIM21 (Addgene, Watertown, USA, 104973) and pHR-LaG16-LaG2 (Addgene, Watertown, USA, 85421) were purchased from Addgene. C-terminal His6-Flag tagged HLTV-αEGFP TRIMbody was generated by subcloning RBCC motif of TRIM21 and LaG16-LaG2 fragment into the HLTV expression vector (BamHⅠ-EcoRⅠ) using ClonExpress MultiS One Step Cloning Kit (Vazyme, Nanjing, China, C113-01). RBCC-LaG16-LaG2 (αEGFP TRIMbody) was then subcloned into a “all-in-one” tetracycline-inducible promoter construct (pTet-on-3G) using the BamHⅠ and EcoRⅠ sites. The LaG16-LaG2 gene was cloned into the pComb3x phagemid and the EGFP gene was cloned into the pET-28a as described above. pUg-EN2-EGFP was made by cloning the EGFP fragment into lentiviral vector pUg-EN2. All new constructs in this study were verified by DNA sequencing. The plasmids psPAX2 and pMD2.G were a kind gift from Shibo Jiang (Fudan University, Shanghai, China). The recombinant vector was transformed into Top10 or Stbl3 competent cell for propagation.
3. Expression and Purification of TRIM21 and TRIMbody
The recombinant vector plasmids were used for transformation of E. coli strain C43(DE3) pLysS cells. A single and freshly transformed colony was added to 4 mL 2× YT medium with 100 µg/mL ampicillin, 34 µg/mL chloromycetin, and 2% (wt/vol) glucose, incubated at 37 °C with vigorous shaking at 250 rpm for 3~4 h, and then transferred into 200 mL of SB medium with 100 µg/mL ampicillin for continued incubation until optical density of the culture at 600 nm reached 0.6~0.8 (after 3~4 h). Next, IPTG (isopropyl-1-thio-β-d-galactopyranoside) was added to a final concentration of 1 mM to induce protein expression, and the culture was further incubated overnight at 22 °C, 250 rpm. Bacteria were collected by centrifugation at 8000 rpm for 10 min and re-suspended in 30 mL Ni-NTA Binding Buffer (0.1 mol/L PBS, 0.5 mol/L NaCl, pH 8.0). The bacteria solution was lysed by sonication and clarified by centrifugation at 8000 rpm for 10 min at 4 °C. The resulting supernatant was further purified using Ni-NTA column (Cytiva, Stockholm, Sweden, 17526802) according to the manufacturer’s protocol. The protein concentration was measured spectrophotometrically, and the degree of protein purity was determined by SDS-PAGE.
4. αEGFP TRIMbody has High Binding Activity to EGFP Protein In Vitro
TRIM21 belongs to the TRIM protein family and consists of a classic N-terminal RBCC motif and C-terminal PRY-SPRY domain. Among them, the RBCC motif can target protein to the proteasome via its E3 ubiquitin ligase activity and PRY-SPRY domain mediates immunoglobulin Fc fragment binding in a pincer-like interaction [
32,
33,
34] (
Figure 1a). To generate a new construct with both protein degradation activity and antibody-binding specificity, we fused the RBCC motif of TRIM21 to a nanobody that can bind to a targeted antigen. The corresponding protein was designated as TRIMbody, and the system was described as TRIMbody-Away. As shown in
Figure 1b, TRIMbody is a multi-domain protein consisting of an N-terminal RING domain with E3 ubiquitin ligase activity, a B-box domain, a coiled-coil dimerization domain, and a C-terminal nanobody fragment which specifically recognizes intracellular proteins of interest.
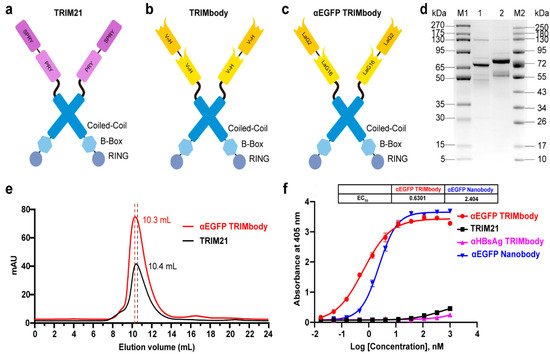
Figure 1. Characterization of TRIM21 and TRIMbody. (a,b) Schematic representation of TRIM21 (a) consisting of an N-terminal RBCC motif and a C-terminal PRY-SPRY domain, and TRIMbody (b) consisting of an RBCC motif and a bispecific nanobody. (c) Schematic representation of the αEGFP TRIMbody consisting of an N-terminal RBCC motif and a C-terminal bispecific anti-EGFP (αEGFP) nanobody (LaG16-LaG2). (d) Coomassie-stained gel shows TRIM21 and αEGFP TRIMbody proteins. Lane 1, protein molecular weight marker 1 (5~270 kDa); Lane 2, TRIM21; Lane 3, αEGFP TRIMbody; Lane 4, protein molecular weight marker 2 (10~250 kDa). (e) Size exclusion chromatography of αEGFP TRIMbody and TRIM21. Protein samples were loaded onto an analytical SuperdexTM 200 Increase 10/300 GL column connected to an FPLC ÄKTA BASIC pH/C system (GE Healthcare). (f) The binding activities of αEGFP TRIMbody, TRIM21, αEGFP Nanobody, and αHBsAg TRIMbody to EGFP were evaluated by ELISA. The EGFP were coated on ELISA plates, and HRP-conjugated anti-Flag antibody was used for detection of binding TRIM21, αEGFP TRIMbody, αEGFP Nanobody, and αHBsAg TRIMbody. Data are shown as mean ± SD.
To test whether TRIMbody could mediate degradation of the target protein, we chose EGFP protein as a proof-of-concept substrate. The previously reported nanobodies against EGFP, LaG16 and LaG2 were selected [
35,
36] and designed for bispecific anti-EGFP (αEGFP) nanobody. Then, an αEGFP TRIMbody was generated that comprised of an RBCC domain and bispecific αEGFP nanobody (
Figure 1c). αEGFP TRIMbody could also be expressed as a soluble form in
E. coli with the help of Lipoyl, as well as TRIM21. By analysis of SDS-PAGE, purified TRIM21 and αEGFP TRIMbody proteins exhibited major bands with molecular weights of 66 and 77 kDa, respectively (
Figure 1d). Next, we examined the oligomeric state of the αEGFP TRIMbody by SEC analysis. The elute volume for αEGFP TRIMbody was determined at 10.3 mL, and 10.4 mL for purified TRIM21 protein, confirming that the αEGFP TRIMbody and TRIM21 have been correctly expressed (
Figure 1e). Further, we measured the binding ability of αEGFP TRIMbody to EGFP by ELISA. Furthermore, a single-chain fragment of G12 antibody that recognizes HBsAg of HBV was used to generated αHBsAg TRIMbody and served as a negative control. We observed that the bispecific anti-EGFP nanobody (LaG16-LaG2) has a EC
50 value of 2.40 nM in EGFP binding, while the αEGFP TRIMbody showed more evident binding activity to EGFP, with an EC
50 value of 0.63 nM (
Figure 1f), which could be due to dimerization of TRIM21 leading to enhanced binding avidity. Remarkably, there was no obvious EGFP binding activity for negative controls.
5. Degradation of Intracellular EGFP by Inducible Expression of αEGFP TRIMbody
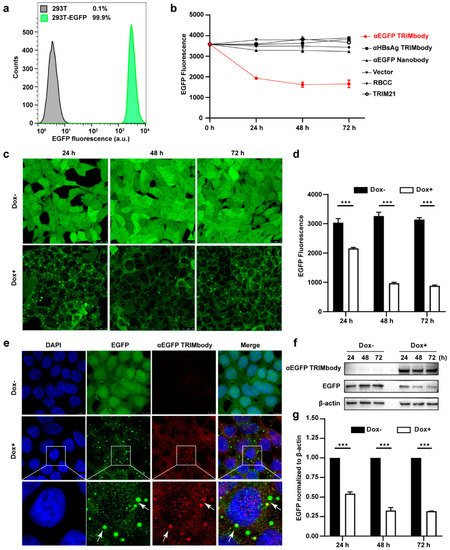
Based on the EGFP binding by αEGFP TRIMbody, we next investigated its target protein degradation function in EGFP-expressing cells. To obtain the stably expressing EGFP cell lines, we performed live cell FACS sorting of 293T cells infected with lentivirus to isolate EGFP-positive cells, and the sorted cells were kept growing and still showed that 99.9% of the population are EGFP-positive at least for five passages, suggesting that expression of EGFP in 293T cells is relatively stable (Figure 2a). Expressing vectors with αEGFP TRIMbody were transiently transfected into EGFP stably expressing 293T cells, and then EGFP fluorescence intensity of cells at the indicated time was determined and analyzed via flow cytometry analysis. Meanwhile, vector, RBCC only, TRIM21 only, αEGFP nanobody, and αHBsAg TRIMbody were used as the controls. We observed that transfection of αEGFP TRIMbody resulted in significant decrease in EGFP fluorescence at 24 and 48 h in a time-dependent manner, but the fluorescence has no significant decrease at 72 h compared with 48 h, which may result from dilution loss of transfected αEGFP TRIMbody plasmid due to transient transfection, leading to insufficient expression of αEGFP TRIMbody protein in cells, or compensatory supply of EGFP protein by constitutive expression of EGFP. In contrast, transfection of RBCC, TRIM21, and αEGFP nanobody showed no decrease of the fluorescence (Figure 2b), suggesting that the RBCC domain and αEGFP nanobody failed to trigger intracellular EGFP degradation. Moreover, we found that αHBsAg TRIMbody, which was confirmed to have no EGFP binding ability in ELISA (Figure 1f), also did not induce intracellular EGFP degradation in EGFP-expressing 293T cells (Figure 2b). The results showed that the combination of the RBCC domain and specific antibody are necessary for degradation of intracellular EGFP.
Figure 2. Degradation of EGFP by αEGFP TRIMbody. (a) Stable expressing EGFP 293T cell lines were analyzed by flow cytometry. At least 15,000 cells were counted for each experiment. Percentages correspond to EGFP-positive cells falling within the gate were drawn. (b) EGFP expressing was decreased in cells after αEGFP TRIMbody transfection. Error bars indicate standard deviations (n = 3). (c) The inducible expression of αEGFP TRIMbody by Dox treatment in stable HEK293T-EGFP cells caused EGFP degradation. Scale bars, 25 μm. (d) Mean fluorescence intensity of EGFP in cells untreated or treated with Dox. Mean fluorescence intensity was measured using flow cytometry and indicated by bar graphs (n = 3 replicates per group). Data represent the mean ± SEM. *** p < 0.001 represents statistical significance. (e) Subcellular localization of αEGFP TRIMbody and EGFP in αEGFP TRIMbody-inducible and EGFP-stable expressing cells. Nuclei were stained with Hoechst 33342; green represents the EGFP protein, red represents the αEGFP TRIMbody tagged with anti-Flag antibody conjugated-Alexa® Fluor 594. White arrows indicate co-localization of EGFP and αEGFP TRIMbody in cytosol of the cells. Scale bars, 7.5 μm. (f,g) The degradation of EGFP was determined by Western blot analysis. Cells were treated with Dox for 72 h and equal amounts of cell lysates (10 μg) were loaded in each well. Through immunoblotting with anti-Flag antibody, the relative optical density of bands on the blots was analyzed by software. Values are the mean ± SEM (n = 3/group). Statistical significance between ligands were determined using a two-way ANOVA test. *** p < 0.001 versus control.
Additionally, to avoid instability and side effects of transient transfection, the inducible expression of αEGFP TRIMbody by Doxycycline (Dox) treatment in stably expressing EGFP cells was constructed using the Tet-On-3G system and further degradation of EGFP was measured by confocal laser scanning microscope or flow cytometry. This system was sensitive to Dox treatment and the optimal Dox concentration was 10 μg/mL. Fluorescence images of cells showed that EGFP was universal and stably expressed and mainly located in the cytoplasm. Notably, αEGFP TRIMbody was not expressed in cells under no Dox treatment, while addition of Dox caused αEGFP TRIMbody expression that co-localized with EGFP (Figure 2e), suggesting that αEGFP TRIMbody can be successfully induced and bound to EGFP via the specificity of the anti-EGFP nanobody. Following Dox addition, EGFP aggregated quickly in cytosol with a spotty pattern at 24 h. In contrast, no significant difference of EGFP was observed without Dox treatment (Figure 2c). Moreover, the quantitative EGFP fluorescence of cells after Dox induction was measured to assess the efficacy of induced αEGFP TRIMbody for EGFP degradation. Cells were treated with Dox and then fixed in 4% paraformaldehyde, and the EGFP fluorescence was analyzed via flow cytometry. The EGFP fluorescence was decreased by 25% at 24 h and had a 67% reduction after 48 and 72 h (Figure 2d). Next, we examined the EGFP and αEGFP TRIMbody protein level after Dox treatment by Western blot analysis and found that αEGFP TRIMbody proteins in cells were induced and kept stably expressed at 24, 48, and 72 h (Figure 2f,g). As for EGFP protein levels, no significant difference was observed in the absence of Dox. By contrast, substantial degradation of EGFP in Dox-treated groups was observed, with 40% reduction at 24 h and 60% decrease at 48 and 72 h after Dox addition (Figure 2g), indicating that intracellular EGFP degradation was subject to Dox induction and αEGFP TRIMbody expression.
6. Dynamic Examination of EGFP Degradation and Accompanying Fluorescent Puncta within 24 h after αEGFP TRIMbody Induction
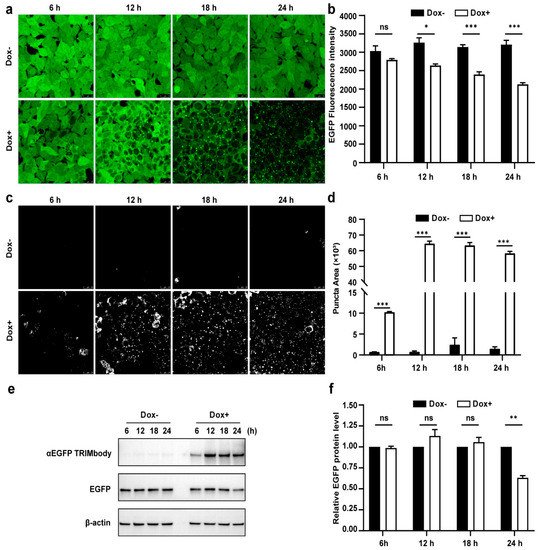
Due to the fact that there was a significant decrease of EGFP protein level at 24 h induction by Dox, we expected that the αEGFP TRIMbody-mediated EGFP degradation process may happen earlier. Therefore, to further characterize the EGFP degradation dynamic pattern and visualize EGFP fluorescence puncta over time upon αEGFP TRIMbody expression, cells were exposed to Dox or vehicle for as long as 24 h and imaged every 6 h using a laser scanning confocal fluorescence microscope (Figure 3a). Meanwhile, we quantified the EGFP fluorescence intensity of cells by flow cytometry analysis (Figure 3b). The punctate EGFP was observed 6 h later by Dox induction and the EGFP fluorescence signal was significantly decreased after 12 h of Dox treatment, reaching maximal reduction of 30% of EGFP fluorescence intensity compared to control at 24 h (Figure 3a,b). Next, EGFP fluorescent puncta was examined from fluorescent images using ImageJ software and we observed it appear as early as 6 h in Dox-treated cells (Figure 3c). Additionally, the puncta area increased dramatically by at least 60-fold compared to controls at 12 and 24 h of Dox treatment (Figure 3d). We speculate that formation of EGFP puncta upon Dox treatment may be due to of EGFP in cytosol through αEGFP TRIMbody induction and recruitment.
Figure 3. Characterization of αEGFP TRIMbody temporal expression pattern. (a) Laser scanning confocal fluorescence microscopy images showed the change in EGFP expression over time with and without Doxycycline. Scale bars are 25 µm. (b) Mean fluorescence intensity of EGFP in cells that were untreated or treated with Dox. Mean fluorescence intensity is measured using flow cytometry and indicated by bar graphs (n = 3 replicates per group). Data represent the mean ± SEM. The ns represents no significance, * represents p < 0.05, and *** represents p < 0.001. (c) EGFP fluorescent puncta was examined from fluorescent images using ImageJ software. (d) Relative EGFP puncta area of autophagosomes or autolysosomes was measured using ImageJ software. Statistical analysis of the puncta area of autophagosomes and autolysosomes per cell were samples from a pool of at least 3 images. Data represent the mean ± SEM. ns represents no significance, *** represents p < 0.001. (e) Western blotting analyzed EGFP and αEGFP TRIMbody levels in the total cell lysate at the indicated times. Equal amounts of cell lysates (10 μg) were loaded in each well and immunoblotted with anti-Flag antibody. (f) Graphs show statistic results from relative optical density of bands on the blots. Values are the mean ± SEM. (n = 3/group). Statistical significance between ligands was determined using a two-way ANOVA test. ** p < 0.01, versus control.
Furthermore, we assessed the EGFP and αEGFP TRIMbody protein level by Western blot analysis and found that EGFP significantly reduced at 24 h of Dox treatment, but not for 6, 12, and 18 h of Dox induction (Figure 3e,f). Meanwhile, αEGFP TRIMbody was successfully induced at 6 h of Dox treatment, reaching to higher levels after adding Dox for 12 to 24 h. We noticed that the EGFP puncta appeared at 6 h of Dox treatment, accompanied with induction of αEGFP TRIMbody, and followed by the reduction of the EGFP fluorescence signal. Finally, the EGFP protein level was detected to decrease at 24 h of induction. These results implied that EGFP degradation happened with the formation of puncta, which may be through αEGFP TRIMbody-mediated intracellular EGFP aggregation, then followed by change of conformation of EGFP, leading to reduction of the fluorescence signal, finally destructed by the RBCC domain-involved degradation pathway. In addition, we performed live-cell imaging on cells and observed the dynamic process of degradation of intracellular EGFP during αEGFP TRIMbody induction.
This entry is adapted from the peer-reviewed paper 10.3390/biom11101512