Heterocyclic nitrogen compounds, including fused 1,5-naphthyridines, have versatile applications in synthetic organic chemistry and play an important role in the field of medicinal chemistry, as many of them have a wide range of biological activities. In this review, a wide range of synthetic protocols for the construction of this scaffold are presented. For example, Friedländer, Skraup, Semmlere-Wolff, and hetero-Diels-Alder, among others, are well known classical synthetic protocols used for the construction of the main 1,5-naphthyridine scaffold. These syntheses are classified according to the nature of the cycle fused to the 1,5-naphthyridine ring: carbocycles, nitrogen heterocycles, oxygen heterocycles, and sulphur heterocycles. In addition, taking into account the aforementioned versatility of these heterocycles, their reactivity is presented as well as their use as a ligand for metal complexes formation. Finally, those fused 1,5-naphthyridines that present biological activity and optical applications, among others, are indicated.
- fused 1,5-naphthyridines
- heterocycle synthesis
- biological activity
- metal complexes.
Heterocyclic nitrogen compounds, including fused 1,5-naphthyridines, have versatile applications in the fields of synthetic organic chemistry and play an important role in the field of medicinal chemistry, as many of them have a wide range of biological activities. In this review, a wide range of synthetic protocols for the construction of this scaffold are presented. For example, Friedländer, Skraup, Semmlere-Wolff, and hetero-Diels-Alder, among others, are well known classical synthetic protocols used for the construction of the main 1,5-naphthyridine scaffold. These syntheses are classified according to the nature of the cycle fused to the 1,5-naphthyridine ring: carbocycles, nitrogen heterocycles, oxygen heterocycles, and sulphur heterocycles.
- Introduction
Several reviews have appeared in the area of naphthyridines [1–4] including some references to fused 1,5-naphthyridines. Among them, in 2005, Ivanov et al. [5] published a generic review on benzo[b]naphthyridines. However, there are no previous reviews that specifically address the chemistry and application of fused 1,5-naphthyridines. For these reasons, to avoid overlapping with previous contributions, our wish in this review is coverage since 2003.
Heterocyclic compounds, such as fused 1,5-naphthyridines, are significantly important in the field of medicinal chemistry, because many of them present a wide variety of biological activities. For example, it was reported that pyronaridine I (Figure 1) has a high activity against Plasmodium falciparum and Plasmodium. vivax [6]. Benzo[b][1,5]naphthyridine II (Figure 1) presented noticeable cytotoxicity against human HL-60 and HeLa cells grown in culture and topoisomerase II inhibition [7]. Moreover, through chemotherapy of solid-tumor-bearing mice, this compound II resulted as potent as amsacrine (m-AMSA) but less toxic towards the host. In 1994, Sliwa et al. showed that benzo[b]1,5-naphthyridine III (Figure 1) presented higher activity against Gram-positive than Gram-negative strains [8]. Years later, 5H-benzo[c][1,5]naphthyridin-6-ones IV (Figure 1) showed poly ADP ribose polymerase (PARP)-1 inhibition and protective effects in rat models of stroke and heart ischemia [9].
Regarding the organization of this review, first of all, the synthesis of 1,5-naphthyridines fused with carbocycles will be addressed, followed by the synthesis of 1,5-naphthyridines fused with nitrogen heterocycles, fused with oxygen heterocycles, and fused with thieno heterocycles. Afterwards, the reactivity of fused 1,5-naphthyridines is classified by N-alkylation, electrophilic substitution reactions (SEAr), nucleophilic substitution reactions (SNAr), oxidations, reductions, side chain modifications, and metal complex formation. Finally, some properties and applications of these heterocycles studied during this period are reviewed.
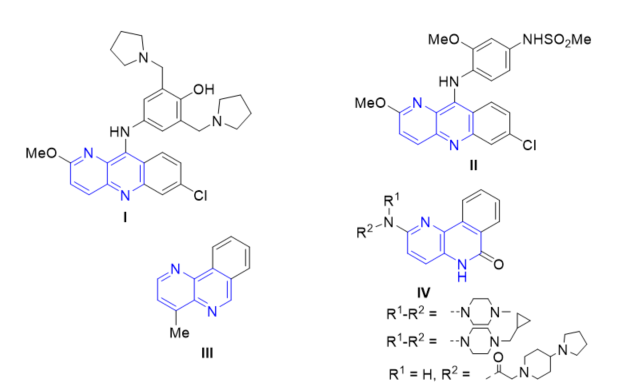
Figure 1. Fused naphthyridines with biological applications.
- Biological Activity of Fused 1,5-Naphthyridines
In the last decade, complex heterocyclic systems containing 1,5-naphthyridines fragments have been synthesized, their reactions have been investigated, and the possibility of their use for the preparation of biologically active compounds have been studied extensively. For example, derivatives of annulated benzoindolo[1,5]naphthyridines attempting to improve memory were described, and these are benzo[b][1,5]naphthyridines which are analogs of inhibitors of neurokinin NK1-receptors [4].
The bromodomain and extra C-terminal (BET) domain family of bromodomains (BRDs) consists of four proteins, each containing two discrete bromodomain “reader” modules which recognize the ɛ-N-acetylation state of specific lysine residues found within histone tails and other proteins [63]. New naphthyridine analogues were synthesized and tested on BET family bromodomains [30]. Compounds 1H-imidazo[4,5-c][1,5]naphthyridin-2(3H)-ones 71b (Scheme 18, vide supra) that possess an isoxazole substituent have as potent inhibitors of the BET bromodomain family with good cell activity and oral pharmacokinetic parameters. Profiling was carried out against the three BET subtypes, as well as in peripheral blood mononuclear cells (PBMCs), where inhibition of cytokine IL-6 was measured after a lipopolysaccharide (LPS) challenge. The fused indenone naphthyridines were more or less equipotent with BRD2, BRD3, BRD4 and PBMC values of pIC50 between 6.1 and 6.8. The best results of pIC50 were for compounds 71bd and 71be (Figure 2).
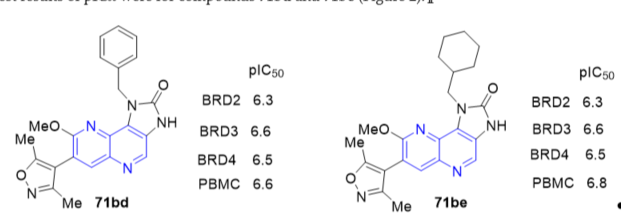
Figure 2. Inhibitory activity of compounds 71bd and 71be on BET bromodomain family.
Benzo[b][1,5]naphthyridine derivatives have been shown to be topoisomerase I (TopI) inhibitors. Compound 162b (7-chloro-2-methoxy-N-(p-tolyl)benzo[b][1,5]naphthyridin-10-amine, Figure 3) was found to display good cytotoxicity and can bind with calf thymus DNA (ct DNA). A relaxation assay indicated that 162b inhibits TopI activity at 100 μM [15]. Compound 162b with methyl substitute at the para-position on the aniline ring displayed good antiproliferative activity with IC50 values of 15.9 and 18.9 µM against K562 and HepG-2 cells respectively in vitro. The ability of compound 162b to interact with ct DNA indicated that nuclear enzymes involved in DNA processing such as TopI might be inhibited. These data suggest that 162b might exert antiproliferative activity through TopI inhibition, and it may be a potential lead compound for the development of benzo[b][1,5]naphthyridines as TopI inhibitors.
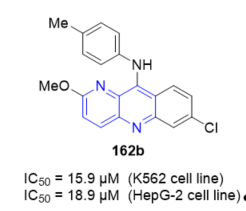
Figure 3. Cytotoxic activity of 162b on K562 and HepG-2 cell lines.
In general, 7H-indeno[2,1-c][1,5]naphthyridines 50 and 51 (Scheme 15, vide supra), and novel 7H-indeno[2,1-c][1,5]naphthyridine-7-ones 193 and 7H-indeno[2,1-c][1,5]naphthyridine-7-ols 194 (Scheme 45, vide supra) exhibited inhibitory effects against TopI mediated relaxation comparable to those observed for the natural inhibitor, camptothecin (CPT). All the prepared derivatives were further subjected to evaluation of their therapeutic efficacy against three different human cancer cell lines: breast (BT20), lung adenocarcinoma (A549), and ovarian carcinoma (SKOV3). These preliminary studies revealed that some of newly synthesized compounds exhibited a significant antiproliferative activity. Indeno[1,5]naphthyridine derivative 51g (Figure 4) showed a high cytotoxic effect in vitro against A549 cell line proliferation with an IC50 of 2.9 ± 0.9 µM. Some of the fluorinated derivatives were the most cytotoxic, showing good enzyme inhibition and relatively low IC50 values. It is worth noting that indeno[1,5]naphthyridine 51d (Figure 4) showed a very high inhibition of TopI activity and the highest cytotoxic effect, with an IC50 of 1.7 ± 0.1 µM, against the A549 cell line in vitro. The interesting biochemical and biological features found for these derivatives provide a promising basis for further development of biologically active fused naphthyridines [24].
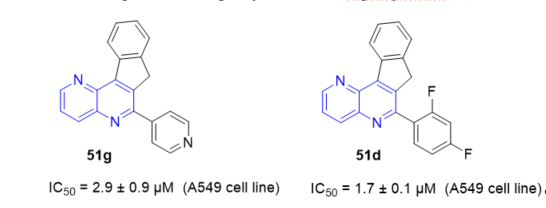
Figure 4. Cytotoxic activity of 51g and 51d on A549 cell line.
The interesting biochemical and biological features found for the above derivatives provide a promising basis for further development of biologically active fused naphthyridines. Thus, tetrahydro[1,5]naphthyridine derivatives fused with heterocycles, such as chromenes 127 and chromen-2-ones 132 and the corresponding tetracyclic chromeno[4,3-b][1,5]naphthyridine 130 derivatives and/or chromeno[4,3-b][1,5]naphthyridin-6-ones 180 (Scheme 47, vide supra), showed activity as inhibitors of TopI [51]. Additionally, the cytotoxic behavior of these compounds has been studied in A549 and SKOV3 cell lines and on noncancerous lung fibroblasts cell line (MRC-5) where, on the last ones, the absence of cytotoxicity was observed.
7-Phenyl-6H-6a,7,12,12a-tetrahydrochromeno[4,3-b][1,5]naphthyridine 127a (Figure 5) showed excellent cytotoxic activity with an IC50 value of 1.03 ± 0.30 µM against the A549 cell line and an IC50 value of 1.75 ± 0.20 µM against the SKOV3 cell line. The obtained results point to these compounds as good antiproliferative candidates.
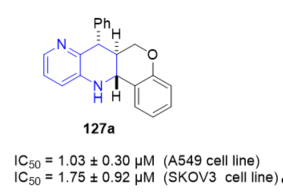
Figure 5. Cytotoxic activity of 127a on A549 and SKOV3 cell lines.
Topoisomerase I enzymatic inhibition of hybrid quinolino[4,3-b][1,5]naphthyridines 110 and 115 (Scheme 47, vide supra) and quinolino[4,3-b][1,5]naphthyridin-6(5H)-ones 111 and 179 (Scheme 47, vide supra) was investigated [45]. The new polycyclic products show excellent-good activity as TopI inhibitors that lead to TopI induced nicking of plasmids. This is consistent with the compounds acting as TopI poisons resulting in the accumulation of trapped cleavage complexes in the DNA. The cytotoxic effect on cell lines A549, SKOV3, and on non-cancerous MRC-5 was also screened. Tetrahydroquinolino[4,3-b][1,5]naphthyridin-6(5H)-one 179 (Figure 6) resulted the most cytotoxic compound with IC50 values of 3.25 ± 0.91 μM and 2.08 ± 1.89 μM against the A549 cell line and the SKOV3 cell line, respectively. Moreover, 5-tosylhexahydroquinolino[4,3-b][1,5]naphthyridine 110a and 5-tosyldihydroquinolino[4,3-b][1,5]naphthyridine 115a (R1 = H) demonstrated to be cytotoxic with IC50 values of 7.25 ± 0.81 μM and 7.34 ± 0.17 μM against the A549 cell line, respectively, and with IC50 values of 8.08 ± 1.39 μM and 8.65 ± 0.57 μM against the SKOV3 cell line, respectively. None of the compounds had cytotoxic effects against non-malignant pulmonary fibroblasts (MRC-5).
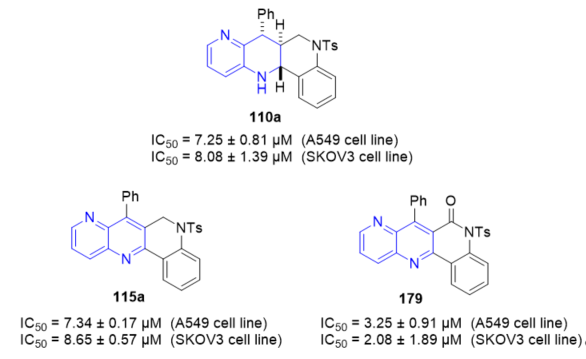
Figure 6. Cytotoxic activity of 110a, 115a, and 179 on A549 and SKOV3 cell lines.
Novel 4,5-dihydro-6λ4pyrrolo[3,2,1-ij][1,5]naphthyridine hydroxy-derivatives including 52 GSK966587 (Figure 7) have been discovered as inhibitors of bacterial type IIA topoisomerases. The compounds showed good potency against Gram-positive and Gram-negative pathogens [27]. The hERG inhibition of the compounds described was generally related to their lipophilicity and basicity and several compounds within this series were identified with hERG inhibition (Figure 7).
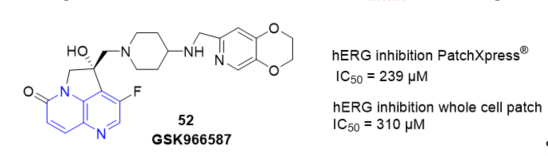
Figure 7. Inhibition of 52 on hERG.
The antitumor activity of fused 1,5-naphthyridines derivatives is closely connected with the intercalation characteristic of many planar ionisable systems. 2-[(Alkylamino)alkyl]-9-methoxy -5-nitro-2,6-dihydroindazolo[4,3-bc][1,5]naphthyridines 94 (Scheme 24, vide supra) represent a new class of aza-acridine derivatives which have noticeable antitumor properties [14]. With enhanced DNA affinity and similar in vitro cytotoxic activity in respect to reference compound (pyrazinamide) PZA, but specially, with a capacity to induce oligonucleosomal DNA fragmentation and apoptotic cell death, not present in PZA. In particular the 9-methoxy-5-nitro-2-[2-(tetrahydro-1H-1-pyrrolyl) ethyl]-2,6-dihydroindazolo[4,3-bc][1,5]naphthyridine 94d, which possesses the most relevant biological characteristics in the series, can be regarded as a new lead in the field of potential anticancer derivatives (Figure 8). Thus, the ability of this compound to early induce oligonucleosomal DNA fragmentation and apoptotic cell death of the hormone-refractory PC-3 prostate cancer cells may be particularly relevant to overcoming drug resistance or sensitize tumor cells to the effects of other antineoplastic agents.
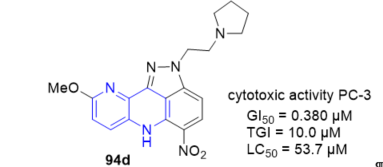
Figure 8. Cytotoxic activity of 94d on PC-3 prostate cancer cells.
To search for a structurally differentiated backup candidate to PF-04691502 (Figure 9), which is currently in phase I/II clinical trials for treating solid tumors, a lead optimization effort was carried out with a tricyclic imidazo[1,5]naphthyridine series. Integration of a structure-based drug design and physical properties-based optimization (Scheme 18, vide supra) yielded PF-04979064, a potent and selective PI3K/mTOR dual kinase inhibitor [29].
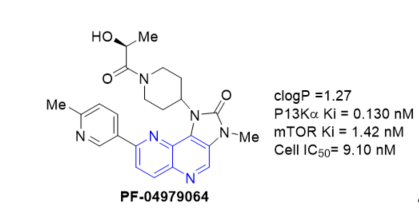
Figure 9. PI3K/mTOR dual kinase inhibition of PF-04979064.
The manuscript discusses the lead optimization for the tricyclic series, which both improved the in vitro potency and addresses a number of absortion, distribution, metabolism, excretion and toxicity (ADMET) properties including high metabolic clearance mediated by both P450 and aldehyde oxidase (AO), poor permeability, and poor solubility. An empirical scaling tool was developed to predict human clearance from in vitro human liver S9 assay data for tricyclic derivatives that were AO substrates.
11,12-Dihydroimidazo[1,2-a]naphtho[1,2-g][1,5]naphthyridine 65 and 7,8-dihydroimidazo [1,2-a]naphtho[2,1-g][1,5]naphthyridine 66 (Scheme 17, vide supra) exhibited in vitro activity comparable to anticancer agent such as amsacrine [28]. Their mechanism of cytotoxicity action was unrelated to poisoning or inhibiting abilities against TopI. On the contrary, must be highlighted a direct intercalation of the drug into DNA by electrophoresis on agarose gel. The tumor cell growth inhibition was assessed on HT-1080 (fibrosarcoma), HT-29 (colon carcinoma), M-21 (skin melanoma), and MCF-7 (breast carcinoma) cells. Compounds 65 and 66 exhibited good values of GI50 ranging from 2.2 to 52 μM (Figure 10). Compound 66 seems to exhibit some specificity towards breast-derived cells such as MCF-7 where it showed a GI50 at least fourfold higher than in any other tumor cell lines (Figure 10).
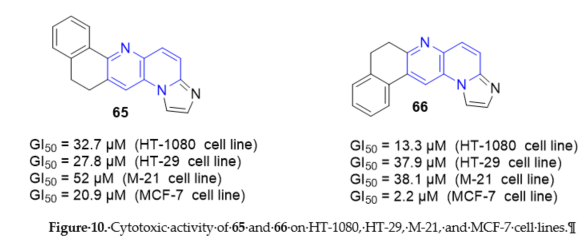
Figure 10. Cytotoxic activity of 65 and 66 on HT-1080, HT-29, M-21, and MCF-7 cell lines.
A library of dimeric benzo[b][1,5]naphthyridines 164 (Figure 11) was synthesized to explore the effect of structurally diverse linkers on PrPSe replication in scrapie-infected neuroblastoma cells [4]. The data suggest that bis-acridine analogs may provide a potent alternative to the acridine-based compound quinacrine which is currently under clinical evaluation for the treatment of prion disease.
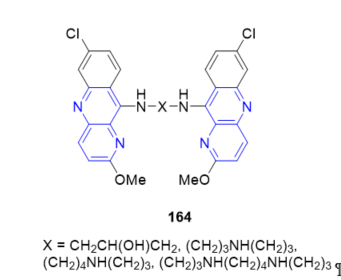
Figure 11. Dimeric benzo[b][1,5]naphthyridines 164 as bis-acridine analogs.
Pyronaridine, 4-((7-chloro-2-methoxybenzo[b][1,5]naphthyridin-10-yl)amino)-2,6-bis (pyrrolidin-1-ylmethyl)phenol 21 (Figure 12), a new Mannich base, schizontocide, originally developed in China and structurally related to the aminoacridine drug quinacrine, is currently undergoing clinical testing. Pyronaridine targets hematin, as demonstrated by its ability to inhibit in vitro β-hematin formation (at a concentration equal to that of chloroquine), to form a complex with hematin with a stoichiometry of 1:2, to enhance hematin-induced red blood cell lysis (but at 1/100 of the chloroquine concentration), and to inhibit glutathione dependent degradation of hematin. Pyronaridine exerted this mechanism of action in situ, based on growth studies of Plasmodium falciparum K1 in culture [64].
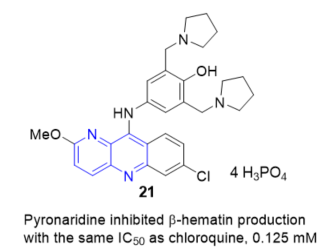
Figure 12. β-Hematin inhibition of 21.
The inhibition of leishmania (LTopIB) and human TopIB (HTopIB) of tetrahydroindeno[1,5]naphthyridines 50 and indeno[1,5]naphthyridines 51 (Scheme 15, vide supra) were studied and their antileishmanial activity on promastigotes and amastigote-infected splenocytes of Leishmania infantum were evaluated [24]. Some of the prepared heterocycles showed selective inhibition of LtopIB, while no inhibition of hTopIB was observed at evaluated conditions. In addition, the cytotoxic effects of newly synthesized compounds were assessed on host murine splenocytes in order to calculate the corresponding selective indexes (SI). Tetrahydro indeno[1,5]naphthyridines 50e and 50h (Figure 13) showed good antileishmanial activity (IC50 values of 0.67 ± 0.06 and 0.54 ± 0.17 µM) with similar activity than the standard drug amphotericin B (0.32 ± 0.05 µM) and even tetrahydro indeno[1,5]naphthyridine 50h showed higher (SI) towards L. Infantum amastigotes. Likewise, in the family of indeno[1,5]naphthyridines 51, compound 51b (Figure 13) showed good antileishmanial activity (IC50 value 0.74 ± 0.08 µM) and higher selective index towards L. Infantum amastigotes than amphotericin.
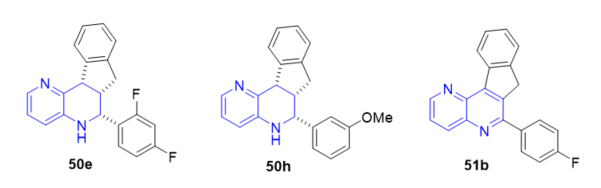
Figure 13. Antileishmanial activity of 50e, 50h, and 51b.
The anti-intestinal nematode activities against Nippostrongylus brazilliensis of a series of benzonaphthyridine derivatives bearing the C=N linkage moiety 187 (Scheme 51, vide supra) were evaluated in vivo by an oral route in male rats [56]. Some of compounds showed significant anti-intestinal nematode activity in a two-day in vivo test in rats. Among these compounds, at concentrations of 10 mg/kg of rat, the compound 7-chloro-2-methoxy- 10-(4-(4’-(1H-indol-5’-yl) methylene)aminophenyl)-amino-benzo[b][1,5]naphthyridine 187n (Figure 14) produced the highest activity against Nippostrongylus brazilliensis, with 80.3% deparasitization. These compounds may find usefulness in the discovery and development of new anti-intestinal drugs.
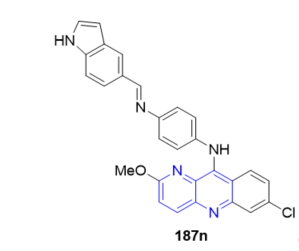
Figure 14. Activity of 187n against Nippostrongylus brazilliensis.
The dimeric indole alkaloid cimiciduphytine 219 (Figure 15) containing the [1,5]naphthyridine fragment and derivatives of eburnane alkaloids 220 (Figure 15) exhibiting hypotensive and pain-relieving activities have been isolated from naturally occurring sources [4]. These compounds are suitable for the treatment of cerebral circulation disturbance.
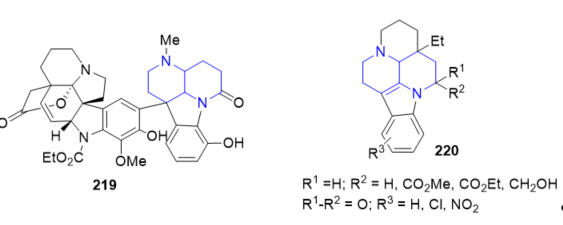
Figure 15. Fused 1,5-naphthyridines with hypotensive and pain-relieving activities.
- Other Applications of Fused 1,5-Naphthyridines
In biological reactions, on the other hand, the NAD+/NADH redox couple, in which the oxidized form (NAD+) with a pyridinium structure is reversibly converted into the reduced form (NADH) with two electrons and one proton, plays a key role in reversible hydride transfer reactions. Transition-metal complexes operating in the same way as the NAD+/NADH system have been reported. Many of these systems present in their structure model ligands that include benzo[b][1,5]naphthyridin-2-yl groups. The electrochemical reduction of 206 (Scheme 1) under aqueous acidic conditions induces formation of the hydrogenated product [Ru(pbnH2)(bpy)2](PF6)2] 221 [10]. The electrochemical reduction of acetone to generate 2-propanol by using complex 206 as a precatalyst with two electrons and H2O as a proton source was described. The key points of this catalytic system are “hydride” generation and transfer, similar to the function of the NAD+/NADH redox couple. Clear evidence of the photochemical and radiolytic formation of 221 with H+ have been reported in Reference [65]. The mechanistic pathways of formation of the NADH-like [Ru(bpy)2(pbnHH)]2+ species from [Ru(bpy)2(pbn)]2+ were studied in an aqueous medium [66] and D2O [67] showing is controlled by the pH of the solution.
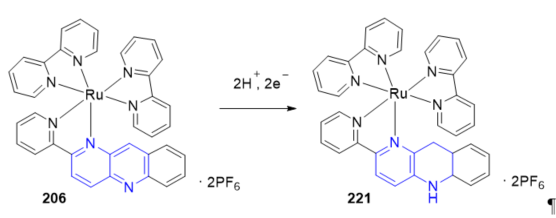
Scheme 1. Formation of the NADH-like species from bpn–ruthenium(II) complex.
On the other hand, on study demonstrated that the introduction of a methyl group of the pyridine moiety in the pbn ligands played a key role in controlling photo-induced NAD+/NADH type hydrogenation reactions by twisting the ligand at a certain level compared with the case of the non-substituted pbn ligand [68].
The use of the multielectron redox reactions of mononuclear metal complexes under visible light irradiation is a fascinating approach to harvesting solar energy. The photoinduced four- and six-electron reduction of [Ru(bpy)(pbn)2](PF6)2 209 and [Ru(pbn)3](PF6)2 210 (Figure 16), with two and three pbn ligands, respectively, under irradiation with visible light (l > 420 nm) conducted in dry CH3CN/TEA (4:1, v/v) resulted in only one-electron reduction of these complexes to give (209)+ and (210)+. The high-efficiency storage of photoinduced two-, four-, and six-electron reducing equivalents in 207 (Scheme 68, vide supra), 209 and 210, respectively, using the NAD+ analogous pbn ligands may provide a new pathway for multiple electron and proton transfer to reaction sites under illumination with visible light [59].
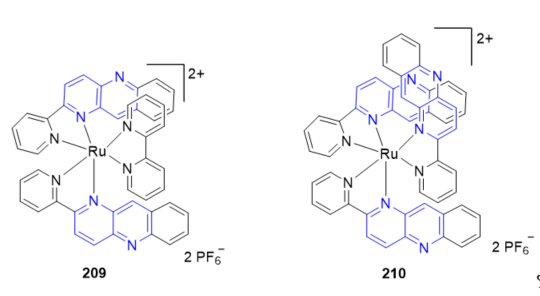
Figure 16. Ruthenium complexes containing fused 1,5-naphthyridines.
The design and synthesis of new catalysts having a capability for photo- and electro-chemical reduction of carbon dioxide (CO2) to produce CO, HCOOH, alcohols, etc., have been pursued to alleviate the global crisis caused by depletion of fossil fuels and the rising atmospheric concentration of CO2. Among the various catalysts, ruthenium complexes may be viable candidates to realize such an innovative function, since they are widely used as catalysts in photo- and electrochemical reduction of CO2 as well as a variety of organic syntheses. The development of a renewable hydride donor is a key process to construct a catalytic system that has the ability to catalyze multi-electron reduction of CO2. Reduction of CO2 using the renewable hydride donor embarks on a new stage of the construction of a sustainable society [69].
An addition of a base to Ru-pbnHH greatly enhanced the hydride donor ability, since [Ru(bpy)2(pbnHH)]2+ 212 reacted with CO2 in the presence of PhCOO_ to give HCOO_ with regeneration of 207 [Ru(bpy)2(pbn)]2+ [70]. [Ru(bpy)2(pbnHH)]2+ 212 was mixed with PhCOO− at −40 °C under CO2 (Scheme 2, R = Ph). The ESI-MS and 1H NMR spectra of the reaction mixture at that temperature displayed 1:1 adduct formation between PhCOO− and [Ru(bpy)2(pbnHH)]2+ 222a (Scheme 69, R = Ph). The association constant (Ka) was 7.8 × 104 M−1.
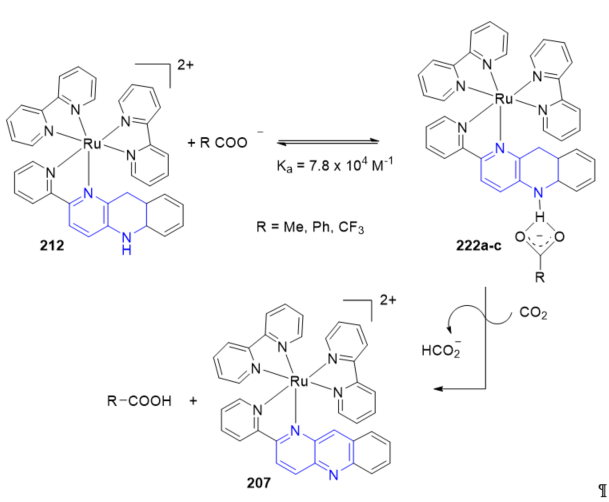
Scheme 2. Photo- and electrochemical reduction of CO2 with bpn–ruthenium complexes.
A drastic difference in the organic hydride transfer reaction converting CO2 to HCO2− using the ruthenium complex containing the NADH model ligand 212 (Scheme 69) was observed by changing the base to either acetate anion (MeCOO−, Scheme 69, R = Me) or trifluoroacetate anion (CF3COO−, (Scheme 70, R = CF3) [71]. The study demonstrated that the choice of the base plays a key role in the CO2 reduction system utilizing 212 through the association of the base to the NH moiety of the NADH model ligand pbnHH (Scheme 69); the difference in basicity between MeCOO− and CF3COO− led to notable accelerating and decelerating effects on the rate of the organic hydride transfer reaction as compared to PhCOO−.
In this context, the electrochemical reduction of CO2 using the catalytic performance of Ru-NAD-type complexes [Ru(tpy)(pbn)(CO)]2+ 216; tpy = 2,2;6,2-terpyridine; pbn = 2-(pyridin-2-yl)benzo[b][1,5]naphthyridine and the Ru-CO-bridged metallacycle 223 was investigated in H2O/CH3CN at room temperature [72]. A controlled-potential electrolysis of 216 and 223 afforded formate (HCOO−) as the main product, under concomitant formation of minor amounts of CO and H2 (Scheme 3).
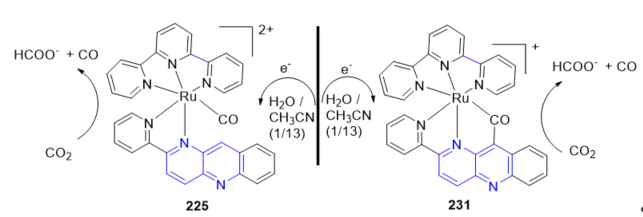
Scheme 3. Electrochemical reduction of CO2 using bpn–ruthenium–NAD-type complexes.
PTNT (poly[thiophene-2,5-diyl-alt-5,10-bis((2-hexyldecyl)oxy)dithieno[3,2-c:3’,2’-h][1,5]- naphthyridine-2,7-diyl]) 149 consists of a tetracyclic fused lactim ring (see Figure 17) that offers many favorable properties, such as improved solid-state packing, high charge carrier mobility, and moderate photoluminescence quantum yields (PLQYs), required for fast optoelectronics. Photophysical properties of PTNT were explored in solution and thin film. Thus, a new strategy to improve the speed of organic light-emitting diodes (OLEDs) using a new class of luminescent polymer with high charge carrier mobilities has been demonstrated [73]. Aqueous nanoparticle dispersions were prepared from PTNT and fullerene blend utilizing chloroform as well as a non-chlorinated and environmentally benign solvent, o-xylene, as the miniemulsion dispersed phase solvent. The nanoparticles (NPs) in the solid-state film were found to coalesce and offered a smooth surface topography upon thermal annealing [74]. Recently, PTNT-conjugated polymer was used as the donor polymer. The preparation of environmentally friendlier polymer solar cell devices [75,76].
Figure 17. Fused 1,5-naphthyridine with OLED properties.
The indeno[2,1-c][1,5]naphthyridine-7-one 193 (Figure 18), which has a 2-naphthyl group, has been used as an optical DNA biosensor to unravel the inhibitory mechanism of human topoisomerase I activity by blocking enzyme—DNA dissociation [77]. This represents the first characterized example of a small molecule drug that inhibits a post-ligation step of catalysis.
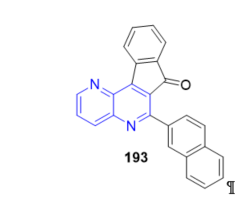
Figure 18. Fused 1,5-naphthyridine as optical DNA biosensor.
The dimmer 98 is quite electron-rich owing to the presence of a 1,2-diaminoethene bridge and is oxidized back to 97 (Scheme 71) within several hours in solution under ambient conditions. Furthermore, 97 possesses a redox-active 1,4-diazabutadiene linkage that is interconvertible with its reduced 1,2-diaminoethene linkage upon treatment of 97 with NaBH4 or PbO2. The dimmer 97 exhibits an intense NIR absorption and narrow HOMO-LUMO gap with a remarkably low reduction potential mainly due to the effective bonding interactions in the LUMO through the 1,4-diazabutadiene linkage. In contrast, the reduced dimmer 98 has high HOMO energy and shows a relative large HOMO-LUMO gap compared to that of 97 [38].
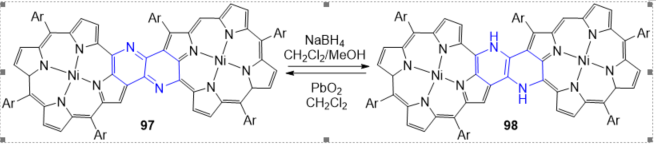
Scheme 4. Redox system of 1,5-naphthyridine-fused porphyrin dimers.
A highly selective and sensitive acridine-base colorimetric sensor 2-((7-chloro-2-methoxybenzo[b]1,5]naphthyridine-10-yl)amino)phenol 224 (NAP, Figure 19) was developed for detection of Cu2+ ions both in aqueous solution and on test papers. Sensor NAP responses to Cu2+ ions by changing its color from yellow to pink, which could be easily observed by the naked eyes [78].
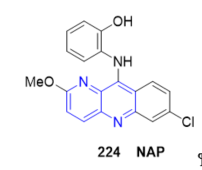
Figure 19. Fused 1,5-naphthyridine as colorimetric sensor for detection of Cu2+ ions.
This entry is adapted from the peer-reviewed paper 10.3390/molecules25153508