Obesity is a multifactorial disorder in which various elements (genetic, host, and environment), play a definite role, even if none of them satisfactorily explains its etiology. A number of neurological comorbidities, such as anxiety and depression, charges the global obesity burden, and evidence suggests the hypothesis that the brain could be the seat of the initial malfunction leading to obesity. The gut microbiome plays an important role in energy homeostasis regulating energy harvesting, fat deposition, as well as feeding behavior and appetite. Dietary patterns, like the Western diet, are known to be a major cause of the obesity epidemic, probably promoting a dysbiotic drift in the gut microbiota.
- obesity
- microbiota
- gut–brain axis
- neurological disorders
- nervous system
- inflammation
1. Introduction
2. Why a Neuropsychological View of Obesity?
3. The Microbiota–Gut–Brain Axis
3.1. Microbiome and Energy Harvest
-
GM influences energy homeostasis by regulating gene expression via complex mechanisms started by SCFAs and monosaccharides [156]. In particular, the commensal microorganisms stimulate monosaccharide cellular uptake [157] and induce lipogenesis by activating the transcription factors carbohydrate response element binding protein (ChREBP) and sterol response element binding protein (SREBP) [155]. Triacylglycerols, produced trought hepatic lipogenesis, are thus sent from the liver to the blood in the form of very low-density lipoprotein and chylomicrons.
-
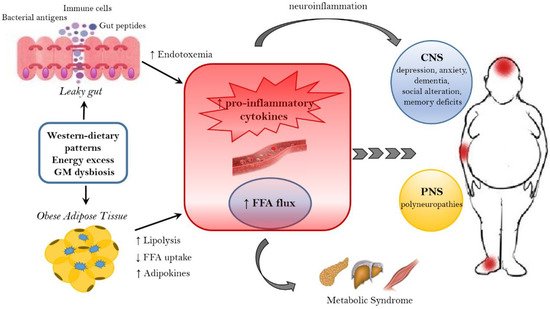
HF diet triggers an increased absorption of bacterial lipopolysaccharide (LPS) (an endotoxin in the cell wall of Gram-negative bacteria) from the gut lumen to the bloodstream inducing low-grade inflammation, by activating B cells or dendritic cells activating and cytokine production [158]. The inflammation could also be stimulated by endotoxemia condition [157]; moreover, also the damaged gut barrier might contribute to this metabolic endotoxaemia [159].
-
GM induces a suppression of angiopoietin-like protein 4 (ANGPTL4) in the intestinal epithelium. ANGPTL4 is a circulating enzyme produced by liver, gut and adipose tissue that inhibits LPL; its suppression provokes an increase of triacylglycerol storage in the adipose tissue [127].
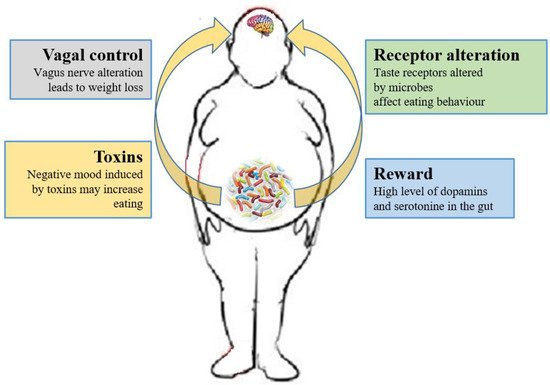
3.2. Microbiome and the Brain
3.3. The Role of Microbiome-Driven Inflammation
This entry is adapted from the peer-reviewed paper 10.3390/nu11010156