Regardless of the pathway, progressive C3 activation results in the formation of the C5-convertases. Correspondingly, the C5-convertases cleave C5 into C5a, an extremely potent inflammatory mediator, and C5b. C5b is the initiator of the terminal step, and, together with the components C6 through C9, assembles the membrane attack complex (MAC), also called C5b-9 [
]. Traditionally, the MAC was found to be formed on Gram-negative bacteria such as Neisseria meningitidis, leading to cell lysis. However, the MAC can also assemble on the surface of other pathogens, erythrocytes, or damaged host cells. Moreover, on host cells, the amount of C9 in the MAC determines the pore size and thereby the function, which ranges from pro-inflammatory effects to cell death [
]. Complement activation also leads to the generation of other effector molecules, such as opsonins (C4b, C4d, C3b, iC3b, and C3dg) and anaphylatoxins (C3a, C5a), which can interact with their respective complement receptors (complement receptors (CR), C3a receptors (C3aR) as well as C5a receptors (C5aR)). To better understand the complement system, it is important to realize that activation can take place in the blood, called the fluid phase, as well as on surfaces, called the solid phase. However, under normal conditions, this system is tightly controlled by regulators present in the blood (fluid-phase regulators) and on cell surfaces (solid-phase regulators) [
]. Examples of solid-phase regulators include membrane cofactor protein (CD46), decay acceleration factor (CD55), the C3b receptor CR1 (CD35), and membrane attack complex-inhibitory protein (CD59), which are widely expressed on human cells. On the other hand, C1-inhibitor, C4b-binding protein (C4bp), Factor H, and Factor I are major fluid-phase regulators present in the blood.
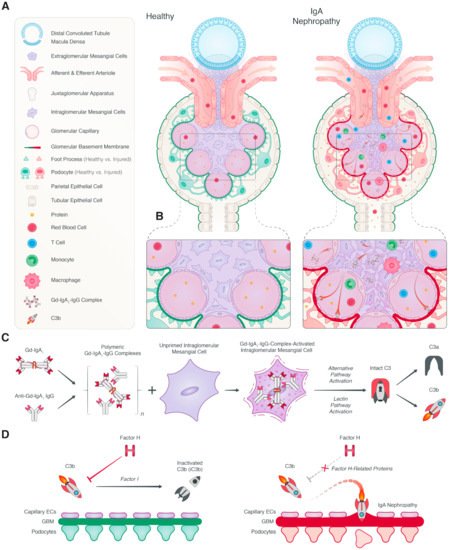
IgAN is the most common form of glomerulonephritis and an important cause of kidney failure [
71]. The diagnosis is confirmed by a kidney biopsy, revealing predominant deposition of IgA1 in the renal mesangium. IgAN is believed to have a multi-hit pathogenesis, namely: genetically determined high circulating levels of galactose-deficient IgA1, subsequent synthesis of antibodies directed against these galactose-deficient IgA, binding of these autoantibodies to IgA1 to form immune complexes, and finally, deposition of the immune complexes in the renal mesangium, leading to immune activation and renal damage [
72]. The presence of complement activation in patients with IgAN was reported almost five decades ago [
73]. However, the relevance of the complement system to the pathophysiology was not immediately recognized. Recent advances have increased our knowledge of the role of the complement system in the pathophysiology of IgAN (
Figure 2). Additionally, these developments have enabled the development of novel therapeutic strategies for IgAN that are currently being tested in clinical trials.
2.1. Local Complement Activation
Very early on, in the initial reports about the disease, complement deposition was already described in renal biopsies of IgAN patients [
73]. These first descriptions of the disease reported mesangial deposition of IgA and C3 in renal biopsies in more than 90% of cases. However, the importance of local complement deposition in IgAN was not recognized until later reports revealed that the extent of C3 deposits in the mesangium correlated with the severity and progression of IgAN [
49,
74,
75,
76,
77]. In these recent studies, glomerular C3 deposition was observed in 71 to 100% of IgAN patients [
78,
79,
80,
81]. Next to glomerular IgA and C3 deposits, properdin and C5b-9 are almost always present, while C1q is typically absent [
49,
73,
82,
83,
84]. Local complement activation in IgAN was therefore thought to result from the alternative pathway. In accordance, early studies demonstrated the ability of IgA to activate the alternative pathway in vitro [
85,
86]. The mechanism behind IgA-induced alternative pathway activation is poorly understood, but the polymerization of IgA is critical. Other proteins of the alternative pathway have also been identified in kidney biopsies of patients with IgAN, including Factor B, Factor H, and the FHRs [
87,
88,
89,
90,
91,
92,
93]. Multiple studies have also investigated the utility of urinary Factor H levels for the assessment of disease activity and prognosis in patients with IgAN [
89,
93,
94,
95]. Surprisingly, urinary levels of Factor H were positively associated with markers of IgAN severity and disease progression. It is noteworthy to mention that because of the structural homology between Factor H and FHRs, it is very well possible that these Factor H assays also detected the FHRs and thereby confound the results [
37]. Proteomic analysis of micro-dissected glomeruli in IgAN biopsies have verified the presence of Factor H, FHR-1, FHR-2, FHR-3, and FHR-5 [
96]. Moreover, FHR-2 and FHR-5 were significantly more abundant in the glomeruli of patients with progressive IgAN compared to non-progressive IgAN. The presence of FHRs in IgAN was first mentioned 20 years ago by Murphy et al., who described glomerular FHR-5 deposits in a range of renal biopsy specimens including IgAN [
97]. Mesangial deposition of FHR-5 was detected in all 20 IgAN cases, and the pattern of FHR-5 deposition was comparable, but not always identical, to that of IgA, C3, and sC5b-9. Recently, increased glomerular staining for FHR-5 was shown to be associated with progressive disease, while a trend was seen for greater FHR1 staining [
88]. In contrast, glomerular Factor H staining was significantly reduced in patients with progressive IgAN in comparison to stable disease. Glomerular FHR5 deposition positively correlated with glomerular staining of C3 activation fragments, C5b-9, and absent Factor H staining.
These results are in line with the hypothesis that FHRs compete with Factor H, thereby amplifying complement activation. No association was seen between glomerular staining for FHR-1 and IgAN severity. Similarly, a Chinese cohort found mesangial staining of FHR-5 in 57.1% of IgAN cases, and FHR-5 deposition was associated with histologic injury [
98]. FHR-5 co-localized and correlated with IgA as well as C3 deposits. IgAN patients with endocapillary hypercellularity and segmental glomerulosclerosis had greater glomerular FHR-5 staining. Interestingly, the authors reported sex differences in glomerular FHR-5 depositions, with greater staining in male IgAN patients. These data indicate that FHR-5 might be a key contributor to complement dysregulation in IgAN (
Table 1). It is important to mention that FHR-5 detection by immunohistochemistry in the study by Medjeral-Thomas et al. and by Guo et al. was achieved by using rabbit polyclonal antibodies against FHR-5 [
88,
98], creating the possibility of cross-reactivity with other FHRs [
37].
Table 1. The role of the Factor H protein family in IgA nephropathy.
| |
Evidence for the Involvement of the Factor H Protein Family in the Pathogenesis of IgA Nephropathy |
| Genetic |
Histologic |
Serologic |
| Factor H |
Genetic variants of Factor H associated with lower plasma levels may contribute to genetic susceptibility to IgAN [99]. |
Glomerular deposition of Factor H staining is reduced in patients with progressive IgAN compared to stable disease. Absence of glomerular Factor H deposition is associated with progressive disease [88]. |
Plasma Factor H levels are not altered in IgAN patients, and these levels are not associated with disease severity, but the plasma FHR-1/Factor H ratio is associated with disease progression [99,100]. |
Factor H-related protein 1
(FHR-1) |
The deletion of complement factor H-related proteins 3 and 1 genes (CFHR3,1Δ) is associated with protection against IgAN [101,102,103,104]. |
Proteomics showed that FHR-1 is more abundant in the glomeruli of IgAN patients compared to controls. Glomerular FHR-1 deposits have also been identified in IgAN, but no association is seen with IgAN severity [88,96]. |
Plasma FHR-1 levels are elevated in IgAN patients compared to healthy controls, and the plasma FHR-1/Factor H ratio is associated with disease progression of the disease [99,100]. |
Factor H-related protein 2
(FHR-2) |
N.D. |
Proteomic analysis revealed that FHR-2 is more abundant in the glomeruli of patients with progressive IgAN compared to non-progressive IgAN [96]. |
N.D. |
Factor H-related protein 3
(FHR-3) |
The deletion of complement factor H-related proteins 3 and 1 genes (CFHR3,1Δ) is associated with protection against IgAN [101,102,103,104]. |
Proteomic analysis demonstrated that FHR-3 is more abundant in the glomeruli of IgAN patients compared to controls [96]. |
N.D. |
Factor H-related protein 4
(FHR-4) |
N.D. |
N.D. |
N.D. |
Factor H-related protein 5
(FHR-5) |
Rare genetic variants in FHR-5 may contribute to the genetic susceptibility to IgAN [105]. |
Glomerular FHR-5 deposits have been identified in IgAN and correlate with C3 and C5b-9 deposits. Increased glomerular staining for FHR-5 is associated with more severe histology and progressive disease [88,96,97,98]. |
Serum FHR-5 levels are higher in IgAN patients compared to healthy controls and are associated with more severe histology, unresponsiveness to immunosuppression, and disease progression [100,106]. |
Although previous studies had shown that the role of the classical pathway is limited in IgAN, little attention had initially been paid to the lectin pathway until the group of Fujita et al. demonstrated glomerular deposition of MBL and MASP-1 in IgAN which co-localized with C3b and C5b-9 deposits [
107]. A follow-up study showed mesangial deposits of MBL, MASP-1, and C4 in over half of the IgAN cases, and also showed that IgA2 co-localized with MBL and MASP-1 in the mesangium of these patients [
108]. Later, additional components of the lectin pathway, such as ficolin-2 deposition, were also demonstrated in IgAN [
78,
109]. In agreement with these results, IgA was shown to induce activation of the lectin pathway in vitro [
16]. Interestingly, lectin pathway presence in renal biopsies is only seen in a subset of IgAN patients [
78,
107,
108]. In the landmark paper by Roos et al., glomerular deposition of Ficolin-2 and MBL was shown to be associated with a higher level of histological damage, demonstrated by increased mesangial and extracapillary proliferation, interstitial infiltration, and glomerular sclerosis, as well as with heavier proteinuria [
78]. Urine levels of MBL and C4d have also been shown to be associated with markers of disease activity and severity in IgAN, and urinary levels of these complement proteins correlate with their respective mesangial deposits [
110,
111]. These findings were further supported by the association of mesangial C4d deposition with disease progression and lower renal survival in IgAN patients [
80,
81,
109,
112]. Espinosa et al. was the first to demonstrate that mesangial C4d staining and absent C1q (indicative of lectin pathway activation) in IgAN patients was associated with progression to kidney failure [
112]. In a follow-up study, they assessed the prognostic value of glomerular C4d staining in IgAN in a larger cohort [
80]. Mesangial C4d deposits were identified in 39% of the 283 patients and C4d-positive staining was an independent risk factor for the development of kidney failure in IgAN. These results had important practical implications, because C4d staining is already routinely used in clinical practice for the diagnosis of antibody-mediated humoral rejection in biopsies from kidney transplant patients [
113]. Various studies have subsequently investigated the use of C4d staining in IgAN as an indicator of disease severity and as a risk factor for kidney outcomes in different geographical populations, stages of chronic kidney disease, and degree of proteinuria [
77,
81,
109,
114]. Recently, a meta-analysis was performed on IgAN studies evaluating the relationship between glomerular C4d deposits and kidney outcomes, and the authors found that glomerular C4d deposition in IgAN was associated with higher histological disease activity, faster decline in eGFR, and kidney failure [
115]. However, C4d deposition in IgAN is not limited to the glomeruli and has also been documented in the vasculature of the kidney. Arteriolar C4d deposits in IgAN are also associated with faster disease progression, and the association with progressive kidney disease was found to be stronger than glomerular C4d deposits [
79]. In accordance, in IgAN, C3 deposition is also routinely found in extraglomerular areas such as in Bowman’s capsule and in the arterioles, and these C3 deposits also seem to be associated with worse outcome [
116].
Glomerular C5b-9 deposition in IgAN was first reported over 3 decades ago by Rauterberg and colleagues [
84]. Terminal pathway activation, as shown by C5b-9, was present in all IgAN cases, but not in controls. Furthermore, mesangial deposits of C5b-9 co-localized with both IgA and C3d deposition. Correspondingly, Medjeral-Thomas et al. reported that mesangial C5b-9 staining significantly correlated with both mesangial C3b/iC3b/C3c and C3d staining [
117]. C5b-9 deposition in the glomeruli has been suggested to contribute to podocyte injury and subsequent proteinuria in IgAN [
118]. Furthermore, decreased expression of CR1 (also known as CD35) on podocytes correlated with glomerular C5b-9 deposition in IgAN. These findings insinuate that reduced CR1 expression perhaps increases the sensitivity of podocytes to complement attack in IgAN [
118]. However, decreased CR1 expression on podocytes is a shared histopathological feature among glomerular diseases and is not specific to IgAN [
119]. In addition to the mesangium, C5b-9 can also be found along the capillary wall in the glomerulus, Bowman’s capsule, the tubular basement membrane, and the vascular wall [
120]. In recent studies, the presence of C5b-9 in IgAN biopsies has been confirmed by proteomics analysis of microdissected glomeruli [
96]. Terminal pathway components were significantly more abundant in IgAN biopsies than in healthy controls, as well as in IgAN cases with progressive disease compared to IgAN with non-progressive disease. Furthermore, terminal pathway components were associated with a higher histological score and lower kidney function. In accordance, multiple studies have found a relationship between C5b-9 staining in IgAN and histological lesions as well as clinical outcomes [
120]. Overall, glomerular C5b-9 deposition in IgAN correlates with the extent of glomerulosclerosis, mesangial expansion, hypercellularity, interstitial inflammation, and fibrosis as well as tubular atrophy, whereas tubular C5b-9 staining is associated with the extent of tubular atrophy, interstitial inflammation, and interstitial fibrosis [
121,
122,
123,
124,
125,
126,
127,
128,
129]. Regarding clinical outcome, glomerular and tubular staining of C5b-9 has been associated with kidney function, proteinuria, and progressive IgAN [
84,
117,
124,
125,
127,
130,
131]. Correspondingly, increased C5aR1 expression has also been reported in renal biopsies of IgAN cases, and C5aR1 staining also correlates with histological injury, proteinuria, and kidney function [
132]. C5aR1 staining in IgAN was mainly found on glomerular mesangial cells, tubular epithelial cells, and interstitial infiltrating cells. Similarly, urine levels of C5a and soluble C5b-9 (sC5b-9) have been found to be associated with markers of disease activity in IgAN, thereby further supporting the significance of the terminal pathway [
93,
132].
2.2. Systemic Complement Activation
In addition to local complement activation in IgAN, systemic complement activation has also been evaluated. Although plasma C3 levels are usually normal, activation fragments of C3 are elevated in some patients and correlate with the levels of IgA-containing immune complexes, histology, and disease progression [
133,
134,
135,
136]. However, most of these studies were performed in the 1980s and 1990s. Proteomics analysis of circulating deglycosylated IgA-immune complexes confirmed the presence of C3 activation fragments, such as iC3b, C3c, and C3dg [
137]. More recently, systemic C3 levels were investigated in 343 IgAN patients [
76]. Only 19% had serum C3 levels below the normal range. However, IgAN patients with decreased C3 levels had higher extents of mesangial C3 deposits in their renal biopsy than those with normal C3 levels. Furthermore, serum C3 levels were significantly associated with progression to kidney failure, but the predictive value of serum C3 was lower than clinical markers such as proteinuria and eGFR. In contrast, a separate study of 496 patients with IgAN, of whom 22% had low levels of C3, reported that serum C3 levels did not associate with disease progression [
138]. Others have suggested that for IgAN, serum IgA1/C3 ratio may be a better marker for disease activity and progression than serum C3 levels alone [
139,
140]. Subsequently, Chen et al. investigated the relationship between the serum galactose-deficient IgA1/C3 ratio and disease progression in 1210 IgAN patients [
141]. The galactose-deficient IgA1/C3 ratio had a much stronger association with disease progression than either marker alone, and the risk of kidney failure increased continuously with the ratio. These findings do not only show the potential of galactose-deficient IgA1/C3 ratios for risk assessment in IgAN, but also suggest that the complement-activating ability of the galactose-deficient IgA1 immune complexes determines disease severity. Terminal pathway activation leading to the generation of C5a and sC5b-9 has also been evaluated in IgAN, although much less extensively. A single study performed by Zwirner et al. found no differences in plasma sC5b-9 levels between patients with IgAN, Henoch–Schonlein purpura, and non-immune kidney disease [
135]. In addition, none of the sC5b-9 values in IgAN patients exceeded the normal range, as defined by levels in the non-immune renal disease group. In a larger Taiwanese cohort, plasma levels of C5a were found to be higher in IgAN patients [
87]. However, these patients were compared to healthy controls and patients with primary focal segmental sclerosis. Interestingly, IgAN patients who received immunosuppression had lower levels of C5a as early as 1 month after treatment.
Serologic evidence of alternative pathway activation (and/or the amplification loop) has also been documented in IgAN. Overall, IgAN patients seem to have higher systemic levels of alternative pathway components, as well as complement regulators [
117,
142]. Plasma levels of Ba, the smaller activation fragment of Factor B, were shown to be increased in IgAN patients compared to healthy controls and patients with primary focal segmental sclerosis [
87]. Additionally, plasma Ba levels positively correlated with plasma levels of C5a levels, as well as weakly (yet statistically significantly) with the degree of proteinuria and impaired renal function. Recent work has investigated circulating levels of the FHRs in IgAN. Plasma levels of FHR-1 were shown to be elevated in Spanish IgAN patients compared to controls, whereas Factor H levels were normal [
99]. In accordance, FHR-1/Factor H ratios were also elevated in IgAN, and the highest FHR-1 levels and FHR-1/Factor H ratios were found in patients with IgAN with disease progression. A separate study confirmed these results and demonstrated that plasma FHR-1 and the plasma FHR-1/Factor H ratio were increased in IgAN and associated with progression of the disease [
100]. In addition, two independent studies showed that serum levels of FHR-5 were significantly higher in IgAN patients than in control patients [
100,
106]. In a British cohort, serum levels of FHR-5 were associated with more severe histology and unresponsiveness to immunosuppression, but not with progressive disease [
100]. In a Chinese cohort, serum levels of FHR-5 were also associated with increased histological injury [
106]. However, in contrast to the British cohort, Zhu et al. did report an association between serum FHR-5 levels and the risk of progressive disease. Whether these differences are due to dissimilar definitions of progressive disease or the consequence of ethnic/geographical differences remains to be determined. Nevertheless, these data, therefore, support the hypothesis that FHR-1 and FHR-5 compete with the regulatory function of Factor H. Factor H tips the balance towards alternative pathway inhibition and reduces the severity of the inflammatory injury, whereas these FHRs amplify alternative pathway activation and thereby stimulate IgAN development and progression of the disease (
Table 1) [
143].
Circulating levels of lectin pathway components have also been linked to IgAN severity. However, this association was complex and U-shaped, indicating that both low and high MBL levels associate with a higher risk, whereas IgAN patients with midrange levels are protected [
144]. MBL deficiency in IgAN patients was associated with 50% loss of kidney function or kidney failure, whereas high levels of MBL (>3540 ng/mL) was associated with various markers of disease severity, including cellular crescents in the kidney biopsy and the degree of proteinuria, although the significance was lost after adjustment for other clinical variables. Furthermore, circulating levels of MBL do not seem to correlate with glomerular MBL deposits in the kidney biopsy [
78]. Plasma levels of other lectin pathway components have also been investigated in IgAN. Circulating levels of ficolin-1, ficolin-2, MASP-1, and MBL-associated protein 2 (MAP-2) were increased in IgAN patients compared to healthy controls, but did not differ between IgAN patients with stable and progressive disease [
117]. MAP-2 (previously MAp19) is an alternative splice product of the MASP-2 gene, and since this truncated form of 19 kDa lacks the serine protease domain, little is known about its function [
145]. Earlier studies also reported systemic C4 activation in IgAN patients. Plasma C4d/C4 ratios, as a marker of C4 activation, were increased on at least one occasion in 28% of the adult IgAN patients [
136]. Unfortunately, these studies have not been repeated since then. It would be especially interesting to see if plasma C4d levels in IgAN patients correlate with the extent of glomerular C4d deposits, since this has been demonstrated for other types of glomerulonephritis [
146]. Initially, serum levels of C4bp were reported to be higher in IgAN patients than controls [
90]. Others were not able to confirm these results, but did find that C4bp levels were higher in IgAN patients with worse prognoses [
142]. Recently, Medjeral-Thomas et al. demonstrated that IgAN patients have reduced levels of MASP-3 compared to healthy controls [
117]. Moreover, reduced MASP-3 levels were associated with the progression of IgAN [
90]. These findings warrant further investigation, since MASP-3 is a vital player in the interaction between LP and AP and could clarify the connection between these two pathways in IgAN [
25].
2.3. Genetic Variants in Complement Genes
Numerous studies support a strong genetic contribution to IgAN, and it was through these genetic studies that the concept of an autoimmune etiology originated [
147]. Genome-wide association studies (GWAS) have revealed that disease susceptibility is greatly impacted by genetic variants in the antigen processing and presentation pathway, as well as the mucosal defense system [
101,
102,
148]. Furthermore, GWAS highlighted the involvement of the complement system in IgAN [
101,
102,
103]. These studies identified a common deletion within the Factor H gene locus as protective against IgAN (
Table 1). This protective deletion results in the loss of the genes for FHR-3 and FHR-1 (CFHR3,1Δ) while leaving the gene for Factor H intact, and each copy of the deletion reduces the risk of IgAN by nearly 40% [
101,
103]. Interestingly, CFHR3,1Δ has been found with a relatively high prevalence, and the population frequency ranges from 0% in East Asians to 20% in Europeans, and up to 50% in certain African populations [
149]. Moreover, CFHR3,1Δ has been associated with a lower risk for the development of AMD and IgAN, whereas it increases the risk for systemic lupus erythematosus (SLE) and aHUS (because of anti-Factor H autoantibodies) [
101,
150,
151,
152]. Fine mapping of the Factor H gene cluster in Chinese cohorts confirmed that CFHR3,1Δ is strongly protective against IgAN [
104]. Furthermore, in IgAN patients, the deletion was associated with a lower prevalence of glomerular segmental sclerosis, tubular atrophy and interstitial fibrosis [
104]. Further mechanistic studies revealed that CFHR3,1Δ in IgAN is associated with reduced mesangial C3 deposition and higher circulating levels of Factor H and C3, together with lower circulating C3a levels [
153,
154]. Recently, CFHR3,1Δ was also shown to be associated with better graft survival in patients who received a kidney transplant for IgAN [
155]. In conclusion, the mechanism behind the protective effect of CFHR3,1Δ in IgAN is thought to arise from the reduced competition of FHRs with Factor H, thereby promoting inhibition rather than activation and accordingly reducing inflammation. In conformity, genetic variants of Factor H associated with lower plasma levels have also been identified in IgAN patients, suggesting that impaired regulation due to Factor H deficiencies could equally increase disease susceptibility [
99]. Rare genetic variants of FHR-5 have also been described in IgAN, and allele frequencies differed significantly from that in controls [
105]. The exact mechanism behind the association of these variants with IgAN remains unclear, but the FHR-5 variants are suggested to have increased binding capacity for C3b [
105].
Genetics have also been utilized to advance the understanding of the lectin pathway in IgAN susceptibility and severity, especially for MBL. In the general population, there is a wide variation in circulating levels of MBL due to common genetic variants in the MBL gene (MBL2) [
156]. The incidence of a MBL deficiency differs among populations, with the highest reported prevalence of more than 60% found in certain South American Indian groups [
157]. The influence of MBL polymorphisms in IgAN was first investigated in a cohort of 77 IgAN patients and 140 controls [
158]. Although no major conclusions could be drawn from this initial study, it is interesting to note that certain allele frequencies were lower in IgAN patients compared to controls. Conversely, Shi et al. found that IgAN patients with an MBL polymorphism in codon 54, which is associated with lower plasma levels, had a worse prognosis [
159]. A separate study of Chinese patients investigated the impact of MBL2 gene polymorphisms on IgAN in a cohort of 749 IgAN patients and 489 controls [
144]. The study found no differences in MBL2 haplotypes between IgAN patients and healthy controls, although a tendency was seen for a lower frequency of the O allele, which leads to a reduction in MBL functionality. These findings would suggest a protective role for low-producing MBL variants. Recently, the impact of MBL2 and ficolin-2 gene (FCN2) polymorphisms on disease progression were explored in over 1000 IgAN patients [
160]. After screening for candidate variants through complete genetic sequencing of MBL2 and FCN2 in a small subset of patients, 7 expression-associated variations were further assessed in the discovery cohort. After adjustment for clinical and pathologic risk factors in multivariate analysis, only one variant in MBL2 (rs1800450) was associated with progression to kidney failure in IgAN patients. Moreover, the association remained significant in their validation cohort. The minor allele of rs1800450 G > A polymorphism was found to be associated with lower plasma levels of MBL, and homozygous IgAN patients had no detectable MBL levels, no glomerular deposition of MBL, increased histological injury as well as an increased risk of disease progression to kidney failure. Overall, the impact of MBL2 variants on IgAN can therefore not be unequivocally defined, since low-producing variants have both been suggested to be detrimental and beneficial.