Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Physiology
Tissue fibrosis is characterized by excessive deposition of extracellular matrix (ECM) components that result from the disruption of regulatory processes responsible for ECM synthesis, deposition, and remodeling. Fibrosis develops in response to a trigger or injury and can occur in nearly all organs of the body.
- fibrosis
- stem cells
1. Introduction
Fibrosis is the excessive accumulation of extracellular matrix (ECM), which leads to impairment of organ function and is consequently associated with high morbidity and mortality. The fibrotic process affects nearly all solid tissues and organs, including the heart, kidney, lung, liver, and skin, and in the majority of cases, this condition results from an ongoing disease (e.g., asthma, hypertension, diabetes, myocardial infarction (MI)) that commonly triggers these tissues [1,2]. Most fibrotic diseases have a relatively well-described etiology, including genetic predisposition, lifestyle, or systemic disorders (Figure 1). Despite showing different clinical features, the majority of fibrotic diseases share common pathological processes characterized by persistent inflammation leading to the production of growth factors, cytokines, and proteolytic enzymes. Altogether, these factors affect myofibroblast differentiation and stimulate excessive deposition of connective tissue components [3].
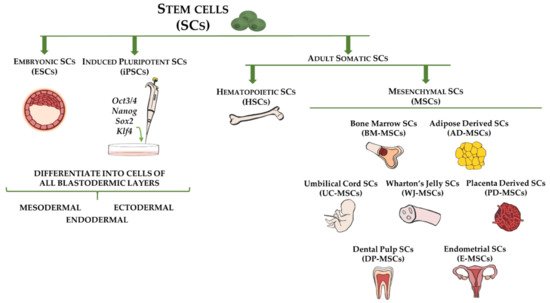
Figure 1. Stem cell types and sources used for cell-based therapy for fibrotic disorders. Embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) are capable of differentiating into cells of all three germ layers (ectoderm, mesoderm, and endoderm). Adult somatic stem cells include hematopoietic stem cells (HSCs), which are multipotent and reside in bone marrow, and mesenchymal stem cells (MSCs), which represent mesodermal progenitors existing in multiple tissues, including bone marrow, adipose tissue, umbilical cord blood, Wharton’s jelly, placenta, dental pulp, and endometrium.
It has been estimated that severe fibrotic disorders are responsible for up to one-third of deaths worldwide [1]. Researchers and clinicians all over the world have worked on developing efficient therapeutic strategies to treat these life-threatening diseases. However, despite constant efforts, there are still no effective therapies for the treatment of fibrosis in any organ (Figure 1). Stem cell-based therapy shows great promise for restoring injured tissues and treating/reversing fibrotic changes. The vast therapeutic potential of this type of therapy in the treatment of neurodegenerative, autoimmune, and genetic disorders has been reported [4,5]. Moreover, clinically relevant studies have included the application of mesenchymal stem cells (MSCs) in a broad range of degenerative, post-injury, and fibrosis-related diseases [6,7,8].
Mesenchymal stem cells are present in various tissues and organs, including bone marrow, adipose tissue, umbilical cord, and endometrium [9,10,11,12]. According to the standard definition, MSCs are fibroblast-like, adherent, clonogenic cells that express a number of surface markers, such as CD29, CD73, CD90, and CD105, and exhibit the capacity to differentiate into adipogenic, chondrogenic, and osteogenic lineages in vitro [13]. The mechanisms through which transplanted MSCs show their therapeutic effects rely on cell differentiation into tissue-specific cell types that replace defective cells or on the secretion of growth factors that lead to an enhanced renewal of target tissue [14,15]. Notably, in vivo studies using rodent models of degenerative neurological diseases have shown that MSCs might contribute to tissue repair by acting as immunomodulatory, neuroprotective, and anti-inflammatory agents [16,17].
In this review, we summarize the results of recent studies regarding the use of stem cells in experimentally induced and clinical cases of fibrosis in multiple organs as well as we present recent patents for stem cell-based therapies in organ fibrosis. We also discuss challenges, limitations, and future perspectives of cell-based therapies that should be considered before conducting large-scale trials.
2. Cellular and Molecular Basis of Tissue Fibrosis
It has been long established that myofibroblasts are the cells responsible for tissue fibrosis [18,19]. Myofibroblasts are heterogeneous cell populations that are defined by the expression of the contractile protein α-smooth muscle actin (αSMA or ACTA2) and the ability to synthesize and release ECM proteins such as collagen (COL) and fibronectin (FN) [18]. A variety of cell types, including resident fibroblasts, vascular pericytes, and bone marrow-derived cells, can be precursors of myofibroblasts [18]. Moreover, in the skin and the lung, epithelial cells can also contribute to the myofibroblast pool as they undergo the epithelial-to-mesenchymal transition (EMT) process [20,21]. Furthermore, as demonstrated in a study by Marangoni et al. [22], subcutaneous adipocytes can transdifferentiate into myofibroblast-like cells upon transforming growth factor beta (TGF)-β stimulation.
The development of fibrotic diseases is associated with abnormal accumulation of myofibroblasts. This aberration leads to excessive deposition of ECM components, which impairs organ structure and function. In healthy tissues, there is a balance between the synthesis and degradation of collagen and other ECM components. These processes are disrupted in fibrotic organs and directed toward ECM accumulation, which is partially evoked by an increased expression of tissue inhibitors of matrix metalloproteinases (TIMPs) versus a decrease in matrix metalloproteinases (MMPs) [23,24]. Indeed, numerous fibrotic-related human liver diseases, including biliary atresia, primary biliary cirrhosis, and primary sclerosing cholangitis, are associated with TIMP-1 and TIMP-2 overexpression [25]. Moreover, membrane-bound matrix metalloproteinase (MT1-MMP)-deficient mice that show a deficiency of membrane-type MMP-14, the metalloproteinase responsible for pericellular proteolysis of ECM, develop progressive fibrosis of the dermis and hair follicles [26].
Although multiple growth factors and pathways contribute to myofibroblast differentiation and EMT process, TGFβ signaling is the major inducer of fibrotic lesions, as has been evident in several in vitro and in vivo studies [27,28]. The activation of TGFβ signaling by overexpression of constitutively active TGFβ receptor type 1 (TGFβRI) is sufficient to induce a fibrotic phenotype characteristic of systemic sclerosis (SS) in mice [29]. In contrast, inhibition of the TGFβ pathway by phosphodiesterase (PDE) inhibitors exert potent antifibrotic effects in human lung fibroblasts [30]. TGFβ signaling is transduced through Smad and non-Smad pathways, such as mitogen-activated protein kinase pathways mediated by extracellular-signal-regulated kinase (ERK), RHO-associated kinase (ROCK), p38 and JUN N-terminal kinase (JNK), and RAC-α serine/threonine-protein kinase pathways [31]. Moreover, TGFβ1 stimulates the secretion of secondary signals, such as connective tissue growth factor (CTGF), plasminogen activator inhibitor 1 (PAI-1), endothelin-1, and NADPH oxidase 4 [32]. These agents show the capacity to mediate and/or mimic the effects of TGFβ1 and ultimately participate in signaling crosstalk, which consequently results in abnormal tissue remodeling and fibrosis in several organs [32].
At present, there is a large amount of data indicating that Wnt and hedgehog signaling, well-described developmental (morphogen) pathways, are potent modulators of fibrotic tissue remodeling across organs [33,34]. Overexpression of Wnt1 and Wnt10b proteins has been observed in human samples from SS, idiopathic pulmonary fibrosis (IPF), and liver cirrhosis [33]. Moreover, it has been shown that β-catenin, the central component of the canonical Wnt signaling, accumulates in the nuclei of fibroblasts from patients with SS and is overexpressed in the skin of mice with bleomycin (BLM)-induced SS. Interestingly, in parallel with the upregulation of Wnt ligands, a significant decrease in the endogenous Wnt antagonist Dickkopf-1 (Dkk-1) was reported [33]. Similarly, other studies demonstrated increased expression of sonic hedgehog (SHH) and the hedgehog transcription factor GLI2 in the skin of patients with SS [35]. At the same time, in the serum of those patients, high SHH concentrations were observed, correlating with the fibrotic burden [36]. Besides the aforementioned key regulators of fibrosis, there is a wide range of well-established factors that stimulate fibrogenesis, such as inflammatory mediators and cells, hypoxia, and DNA methylation of antifibrotic genes [37,38,39]. Importantly, it must be highlighted that tissue fibrosis is a complex process that involves the action of many factors and the activation of multiple signaling cascades that cross-react at multiple levels, and consequently, this condition presents a challenge in the development of novel antifibrotic approaches.
This entry is adapted from the peer-reviewed paper 10.3390/life11101068
This entry is offline, you can click here to edit this entry!