Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Pharmacology & Pharmacy
Oxycodone is a widely used opioid for the management of chronic pain. Analgesic effects observed following the administration of oxycodone are mediated mostly by agonistic effects on the μ-opioid receptor. Wide inter-subject variability observed in oxycodone efficacy could be explained by polymorphisms in the gene coding for the μ-opioid receptor (OPRM1).
- oxycodone
- pharmacogenetics
- CYP2D6
- OPRM1
- COMT
- efficacy
1. Introduction
Drug overdose deaths involving prescription opioids (natural and semi-synthetic opioids and methadone) rose from 3442 in 1999 to 14,139 in 2019, reaching a peak of 17,209 deaths in 2017 [1]. Oxycodone is a semi-synthetic opioid used for pain management, accounting for approximately 17 million prescriptions in 2018 [2]. In 2016–2017, oxycodone prescriptions represented approximately 18% of all pain management prescriptions following an emergency room visit in the United States [3]. Recently, the role of genetic polymorphisms associated with proteins involved in the pharmacokinetics and pharmacodynamics of oxycodone was reviewed by the Clinical Pharmacogenetics Implementation Consortium (CPIC), which concluded that current evidence was insufficient to make firm clinical recommendations—in the form of a clinical guideline—regarding genetic testing for oxycodone dose adjustment [4]. Some study results have clearly demonstrated and supported the value of genetic testing, though others have not found clinical value in genetic testing to improve pain management with oxycodone. This is especially the case for polymorphisms associated with CYP2D6 and the formation of oxymorphone—an active metabolite of oxycodone—and for polymorphisms linked to the opioid receptor mu 1 gene (OPMR1) [5].
2. Pharmacokinetic Considerations
2.1. CYP2D6 Activities, Polymorphisms, and Expression (GRADE High Quality ++++)
The cytochrome P450 (CYP450) superfamily is comprised of several heme-containing monooxygenases involved in the phase I metabolism of ~75% of small molecules and is a crucial part of our current pharmacological armamentarium. In humans, this superfamily of enzymes is encoded by 57 genes from 18 families [6]. One gene, CYP2D6, codes for an enzyme involved in the metabolism of eicosanoids, drugs, and foreign chemicals [7][8][9][10]. CYP2D6 is a highly polymorphic gene, and more than 100 variants and subvariants have been identified [11]. CYP2D6 substrates are typically lipophilic bases with an aromatic ring and a nitrogen atom that are protonated at physiological pH. Substrate binding is then followed by oxidation 5–7 Å, apart from the nitrogen moiety [12][13]. CYP2D6 is responsible for the clearance of at least 20% of the compounds in current clinical use, including antiarrhythmics, antidepressants, antipsychotics, β-blockers, and analgesics [8][9][14].
As CYP2D6 allelic variants influence protein expression and activity, CYP2D6 polymorphisms affect the functional capability of the enzyme to metabolize CYP2D6 substrate drugs. Allele combinations can produce poor metabolizers (PMs: non-functioning CYP2D6), intermediate metabolizers (IMs: low-functioning CYP2D6), normal metabolizers (NMs: normal functioning CYP2D6), and ultra-rapid metabolizers (UMs: high-functioning CYP2D6, mostly due to gene duplication). In a large genetic testing study of 104,509 deidentified patient samples, it was extrapolated that in the population, an estimated 5.7% of individuals would be PMs, 10.7% would be IMs, and 2.2% would be UMs, with 81% NMs [15]. However, there are differences in CYP2D6 allele frequencies in distinct racial and ethnic populations. For example, the CYP2D6*4 allele frequency appears higher in Caucasians, CYP2D6*10 in East Asians, CYP2D6*41 and duplication/multiplication of active alleles in those of Middle Eastern descent, CYP2D6*17 in Black Africans, and CYP2D6*29 in African Americans [16][17][18].
CYP2D6 is highly and selectively expressed in the human liver. Few tissues express CYP2D6 and recent studies demonstrated that when using an absolute protein assay and LC-MSMS technique, the protein could not be detected in various regions of the human small intestine [19][20]. In 1987, Fonne-Pfister et al. demonstrated the expression of CYP2D6 in the human brain [21]. More than 10 years later, dextromethorphan—a well-known CYP2D6 probe substrate—to dextrorphan metabolism was demonstrated in human brain microsomes [22]. CYP2D6 expression has also been described in various brain regions and cell types including the cerebral cortex, hippocampus, cerebellum, basal glia, midbrain, thalamus, and glial cells [23][24][25][26][27]. CYP2D6 is also involved in the metabolism of tyramine to dopamine, in the regeneration of serotonin from 5-methoxytryptamine, and in behavioral traits and psychopathology [28][29][30][31][32].
2.2. Oxycodone Metabolism in Humans (PK and PGx; GRADE High Quality ++++)
Drug metabolism studies have characterized the extensive biotransformation of oxycodone in humans. Oxycodone is N-demethylated to noroxycodone and O-demethylated to oxymorphone; both undergo subsequent major metabolism by either N-demethylation (noroxycodone into noroxymorphone) or glucuronidation (oxymorphone into oxymorphone 3-glucuronide) [33][34]. In vitro studies conducted with human liver microsomes and genetically-engineered recombinant drug-metabolizing enzyme products have demonstrated that CYP3A4 and CYP2D6 isoforms mediate most of oxycodone metabolism [33][35]. A small amount is converted to α/β oxycodol via 6-keto reduction and a small fraction is also directly converted to a glucuronide conjugate (Figure 1) [36].
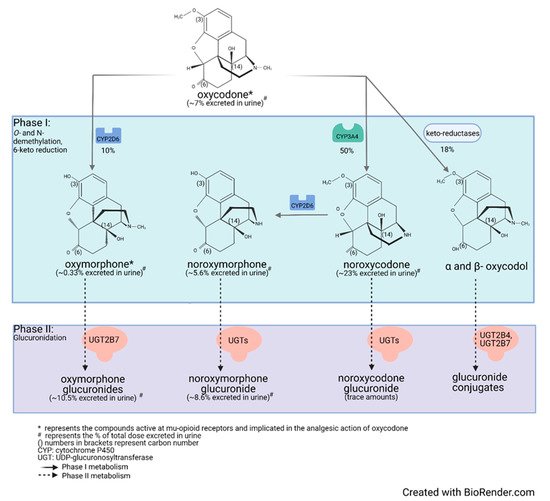
Figure 1. Detailed illustration of the elimination pathways of oxycodone.
Lalovic et al., using human liver microsomes, showed that CYP3A4 was the highest-affinity enzyme involved in the N-demethylation of oxycodone to noroxycodone with a mean Km of 600 ± 119 μM [33]; co-incubation with the CYP3A4 inhibitor ketoconazole decreased formation of noroxycodone by more than 90%. During the O-demethylation of oxycodone to oxymorphone, CYP2D6 was found to have the highest affinity, with a Km of 130 ± 33 μM [33]. Other enzymes contributed to less than 8% for the N-demethylation and 10–26% for the O-demethylation pathways, respectively. N-Demethylation activity in intestinal microsomes was 20–50% that of the liver microsomes, while the O-demethylation was negligible, suggesting that the liver may be primarily responsible for first-pass metabolism, especially for the O-demethylation [33]. These results are in agreement with CYP3A4 and CYP2D6 protein levels measured in the human small intestine [19][37]. Romand et al. confirmed the important roles of CYP3A4 and CYP2D6 in the phase I transformation of oxycodone, while implicating UGT2B7 (Km: 762 ± 153 μM) and to a lesser extent UGT2B4 (Km: 2454 ± 497 μM) in the phase II metabolism of oxycodone [36].
Human mass-balance and pharmacokinetic studies confirmed that oxycodone is extensively metabolized in humans with only 10% of the drug excreted unchanged in urine [38][39][40]. In agreement with in vitro studies, oral administration of oxycodone showed that CYP3A4 is involved in the major metabolic clearance pathway with about 50% of the drug being N-demethylated to noroxycodone, while about 10% is O-demethylated by CYP2D6 to oxymorphone [40][41][42][43]. Following oral administration of oxycodone (15 mg single-dose immediate-release formulation) in 10 healthy individuals, (one CYP2D6 PM, one CYP2D6 UM, eight CYP2D6 activity NMs), average peak plasma concentrations of oxycodone reached approximately 30 ng/mL, while oxymorphone peak plasma concentrations were about 0.7 ng/mL [44][45][46][47]. In plasma of CYP2D6 NMs, the oxycodone-to-oxymorphone ratio could therefore be estimated at ~43:1 (Table 1) [45][46][47]. Under similar conditions, noroxycodone concentrations reached about 20 ng/mL [44][45][46][47].
Table 1. Change in oxycodone:oxymorphone ratio depending on patient genotype or use of CYP450-inhibitor drugs.
| CYP2D6 Activity | Oxycodone/Oxymorphone Concentration Ratio in Plasma | Oxycodone/Oxymorphone Free-Drug Concentration Ratio in Plasma | Oxycodone/Oxymorphone Free-Drug Concentration Ratio in the Brain * | Oxycodone/Oxymorphone Relative Contribution to μ-Opioid Receptor-Binding Considering the Free-Drug Concentration Ratio in the Brain ** |
|---|---|---|---|---|
| UM CYP2D6 | 32:1 | 19:1 | 57:1 | 0.6:1 |
| NM CYP2D6 | 43:1 | 26:1 | 78:1 | 0.8:1 |
| PM CYP2D6 | 300:1 | 180:1 | 540:1 | 5.4:1 |
| With potent CYP2D6 inhibitors | 110:1 | 66:1 | 198:1 | 2:1 |
| With potent CYP3A4 inhibitors | 56:1 to 21:1 | 34:1 to 13:1 | 102:1 to 39:1 | 1:1 to 0.4:1 |
CYP2D6: cytochrome P450 2D6; UM: ultra-rapid metabolizer; NM: normal metabolizer; PM: poor metabolizer; CYP3A4: cytochrome P450 3A4; *: assuming a conservative brain-to-blood unbound concentration ratio of 3.0 for oxycodone and of 1.0 for oxymorphone; **: assuming a conservative brain-to-blood unbound concentration ratio of 3.0 for oxycodone and of 1.0 for oxymorphone and a potency ratio of 1:100 for oxycodone vs. oxymorphone.
Valuable information on the relative contributions of CYP2D6 and CYP3As to the disposition of oxycodone can also be obtained from pharmacogenomic and drug–drug interactions studies. Studies performed in individuals with multiple copies of CYP2D6—which translates to an UM phenotype—have shown that average plasma levels of oxycodone are reduced while average concentrations of oxymorphone are increased (relative to NMs). The oxycodone-to-oxymorphone plasma-concentration ratio was then estimated to be ~32:1 in UMs following oral administration (Table 1) [45][46][47].
In patients and healthy volunteers with non-functional CYP2D6 (PMs), the oxycodone-to-oxymorphone ratio is increased to approximately 300:1 (Table 1) [47][48]. This increased ratio is mostly explained by a significant decrease in oxymorphone plasma concentrations, as the overall contribution of CYP2D6 to the total clearance of oxycodone is limited. Still, a nominal amount of oxymorphone is detected in the plasma of these individuals, suggesting that an isoform other than CYP2D6 could also mediate the formation of oxymorphone. Similar results were obtained during in vitro drug metabolism studies [33]. Notably, much lower ratios of oxycodone/oxymorphone were observed in patients from all phenotypic groups following the administration of oxycodone intravenously [49]. This observation can be explained by a route of administration-dependent difference in the relative contributions of CYP3A4 and CYP2D6 to the clearance of oxycodone, as CYP2D6 expression is limited in the intestinal tissue [20][50][51][52]. Hence, the relative contribution of CYP2D6 to the total clearance of oxycodone appears increased following intravenous administration.
Drug interactions studies have been conducted with potent CYP2D6 inhibitors. Heiskanen et al. and Sirhan-Daneau et al. conducted studies in normal CYP2D6 metabolizers and demonstrated—using quinidine as a non-competitive and potent inhibitor of CYP2D6—that formation of oxymorphone was almost completely impeded under potent CYP2D6 inhibition [45][46][53][54]. However, using sensitive LC-MS/MS assays, limited amounts of oxymorphone could still be detected in their plasma [53]. Similar results were obtained by Lemberg et al. using paroxetine as a mechanism-based inhibitor of CYP2D6 [55]. Under potent inhibition of CYP2D6, the oxycodone-to-oxymorphone plasma-concentration ratio is nearly ~110:1 (Table 1).
As mentioned previously, CYP3A4 is the major enzyme involved in the disposition of oxycodone. Inhibition of CYP3A4, using inhibitory agents such as itraconazole or ketoconazole, is associated with increases in both oxycodone and also oxymorphone levels. As CYP3A4 is inhibited, more oxycodone is available for its metabolism by CYP2D6 into oxymorphone. Under conditions of CYP3A4 inhibition, the oxycodone to oxymorphone plasma concentration ratios ranged from 56:1 to 21:1 (Table 1) [56][57][58].
2.3. Oxycodone Distribution and Protein Binding (PK; GRADE Low Quality ++−−)
Average plasma protein binding for oxycodone is 45%, while that for oxymorphone is 11%. Hence, for all situations described above, the relative oxycodone/oxymorphone ratios while considering free-drug concentrations in the plasma [(Cavg oxycodone × Fu)/(Cavg oxymorphone × Fu)] should be decreased by about 40% (Table 1).
Drug distribution studies have demonstrated that both oxycodone and oxymorphone can cross the blood–brain barrier. First, Bostrom et al. reported in two different studies that the brain-to-blood unbound concentration ratio of oxycodone was 3.0–6.0 [59][60]. Then, Zasshi et al. also reported that the brain-to-blood unbound concentration ratio for oxycodone was 3.0 [61]. In regards to oxymorphone, Sadiq et al. reported that the brain-to-blood unbound concentration ratio for oxymorphone was 1.9 [62]. In contrast, Zasshi et al. and Lalovic et al. reported that the oxymorphone brain-to-blood unbound concentration ratio was 0.26 to 0.3 [33][43][61]. However, the discrepancy between these results could not be explained. Assuming a conservative brain-to-blood unbound concentration ratio of 3.0 for oxycodone and an average of 1.0 for oxymorphone, new oxycodone-to-oxymorphone ratios can be estimated (Table 1); noroxycodone and noroxymorphone do not have good brain penetration [43].
2.4. Oxymorphone Pharmacokinetics Following Direct Oxymorphone Administration (PK; GRADE Moderate Quality +++−)
Oxymorphone is a known potent μ-opioid receptor agonist which may contribute, totally or partially, to the overall analgesic effects observed following oxycodone oral administration [63]. Oxymorphone can be administered directly as an active drug to humans and its pharmacokinetics and pharmacodynamics can be appreciated and compared to its pharmacokinetics/pharmacodynamic relationship observed following oxycodone administration.
The bioavailability of oral oxymorphone is about 10% following an important first-pass metabolism. Oxymorphone undergoes extensive conjugation mediated mostly by UGT2B7 to produce its primary metabolite, oxymorphone 3-glucuronide [64][65]. To a lesser extent, it also undergoes reduction of the 6-ketone group to form 6-hydroxy-oxymorphone. After administration of a single 10 mg oxymorphone immediate-release oral dose, 1.9% was eliminated in urine as free oxymorphone, 44.1% as conjugated oxymorphone 3-glucuronide, 0.3% as 6-hydroxy-oxymorphone, and 2.6% as conjugated 6-hydroxy-oxymorphone [34]. Note that in contrast to other opioids, the N-demethylation of oxymorphone into noroxymorphone is very limited in humans [34].
Following the oral administration of a single extended-release 10 mg dose of oxymorphone, peak plasma concentrations of oxymorphone, 6-hydroxy-oxymorphone, and oxymorphone 3-glucuronide reached 0.65 ng/mL, 0.37 ng/mL, and 112 ng/mL, respectively [66]. Plasma levels of oxymorphone observed under these conditions were similar to plasma concentrations observed following administration of a single 15 mg immediate-release oxycodone dose (0.7 ng/mL) [45][46][54]. This observation strongly suggests that oxymorphone plasma levels of 0.5 to 1.0 ng/mL mediate clinically relevant analgesic effects and likely contribute significantly to oxycodone efficacy.
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics13091466
References
- National Institute on Drug Abuse. Overdose Death Rates. Available online: https://www.drugabuse.gov/drug-topics/trends-statistics/overdose-death-rates (accessed on 22 February 2021).
- Kane, S. Oxycodone, ClinCalc DrugStats Database, Version. Available online: https://clincalc.com/DrugStats/Drugs/Oxycodone (accessed on 22 February 2021).
- Rui, P.; Santo, L.; Ashman, J.J. Trends in opioids prescribed at discharge from emergency departments among adults: United States 2006–2017. Nat. Health Stat. Rep. 2020, 135, 1–12. Available online: https://www.cdc.gov/nchs/data/nhsr/nhsr135-508 (accessed on 22 February 2021).
- Crews, K.R.; Monte, A.A.; Huddart, R.; Caudle, K.E.; Kharasch, E.D.; Gaedigk, A.; Dunnenberger, H.M.; Leeder, J.S.; Callaghan, J.T.; Samer, C.F.; et al. Clinical Pharmacogenetics Implementation Consortium Guideline for CYP2D6, OPRM1, and COMT Genotypes and Select Opioid Therapy. Clin. Pharmacol. Ther. 2021.
- Ellis, C.R.; Kruhlak, N.L.; Kim, M.T.; Hawkins, E.G.; Stavitskaya, L. Predicting opioid receptor binding affinity of pharmacologically unclassified designer substances using molecular docking. PLoS ONE 2018, 13, e0197734.
- Wang, B.; Yang, L.-P.; Zhang, X.-Z.; Huang, S.-Q.; Bartlam, M.; Zhou, S.-F. New insights into the structural characteristics and functional relevance of the human cytochrome P450 2D6 enzyme. Drug Metab. Rev. 2009, 41, 573–643.
- Snider, N.T.; Sikora, M.J.; Sridar, C.; Feuerstein, T.J.; Rae, J.M.; Hollenberg, P.F. The endocannabinoid anandamide is a substrate for the human polymorphic cytochrome P450 2D. J. Pharmacol. Exp. Ther. 2008, 327, 538–545.
- Bertilsson, L.; Dahl, M.-L.; Dalén, P.; Al-Shurbaji, A. Molecular genetics of CYP2D6: Clinical relevance with focus on psychotropic drugs. Br. J. Clin. Pharmacol. 2002, 53, 111–122.
- Haufroid, V.; Hantson, P. CYP2D6 genetic polymorphisms and their relevance for poisoning due to amfetamines, opioid analgesics and antidepressants. Clin. Toxicol. 2015, 53, 501–510.
- Shen, H.W.; Jiang, X.L.; Winter, J.C.; Yu, A.M. Psychedelic 5-methoxy-N,N-dimethyltryptamine: Metabolism, pharmacokinetics, drug interactions, and pharmacological actions. Curr. Drug. Metab. 2010, 11, 659–666.
- The Pharmacogene Variation PharmVar Consortium. CYP2D6 cytochrome P450 Family 2 Subfamily D Member. Available online: https://www.pharmvar.org/gene/CYP2D6 (accessed on 16 September 2016).
- Maréchal, J.D.; Kemp, C.A.; Roberts, G.C.; Paine, M.J.; Wolf, C.R.; Sutcliffe, M.J. Insights into drug metabolism by cytochromes P450 from modelling studies of CYP2D6-drug interactions. Br. J. Pharm. 2008, 153, S82–S89.
- Kapelyukh, Y.; Wolf, R. CYP2D6 substrates and drug metabolism. In CYP2D6: Genetics, Pharmacology and Clinical Relevance; Future Medicine Ltd.: London, UK, 2014; pp. 80–100.
- Anzenbacher, P.; Anzenbacherová, E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol. Life Sci. 2001, 58, 737–747.
- Del Tredici, A.L.; Malhotra, A.; Dedek, M.; Espin, F.; Roach, D.; Zhu, G.D.; Voland, J.; Moreno, T.A. Frequency of CYP2D6 Alleles Including Structural Variants in the United States. Front. Pharm. 2018, 9, 305.
- Byeon, J.Y.; Kim, Y.H.; Lee, C.M.; Kim, S.H.; Chae, W.K.; Jung, E.H.; Choi, C.I.; Jang, C.G.; Lee, S.Y.; Bae, J.W.; et al. CYP2D6 allele frequencies in Korean population, comparison with East Asian, Caucasian and African populations, and the comparison of metabolic activity of CYP2D6 genotypes. Arch. Pharm. Res. 2018, 41, 921–930.
- LLerena, A.; Naranjo, M.E.; Rodrigues-Soares, F.; Penas, L.E.M.; Fariñas, H.; Tarazona-Santos, E. Interethnic variability of CYP2D6 alleles and of predicted and measured metabolic phenotypes across world populations. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1569–1583.
- Oscarson, M.; Hidestrand, M.; Johansson, I.; Ingelman-Sundberg, M. A Combination of Mutations in the CYP2D6*17 (CYP2D6Z) Allele Causes Alterations in Enzyme Function. Mol. Pharmacol. 1997, 52, 1034–1040.
- Grangeon, A.; Clermont, V.; Barama, A.; Gaudette, F.; Turgeon, J.; Michaud, V. Development and validation of an absolute protein assay for the simultaneous quantification of fourteen CYP450s in human microsomes by HPLC-MS/MS-based targeted proteomics. J. Pharm. Biomed. Anal. 2019, 173, 96–107.
- Clermont, V.; Grangeon, A.; Barama, A.; Turgeon, J.; Lallier, M.; Malaise, J.; Michaud, V. Activity and mRNA expression levels of selected cytochromes P450 in various sections of the human small intestine. Br. J. Clin. Pharm. 2019, 85, 1367–1377.
- Fonne-Pfister, R.; Bargetzi, M.J.; Meyer, U.A. MPTP, the neurotoxin inducing Parkinson’s disease, is a potent competitive inhibitor of human and rat cytochrome P450 isozymes (P450bufI, P450db1) catalyzing debrisoquine 4-hydroxylation. Biochem. Biophys. Res. Commun. 1987, 148, 1144–1150.
- Gilham, D.E.; Cairns, W.; Paine, M.J.; Modi, S.; Poulsom, R.; Roberts, G.C.; Wolf, C.R. Metabolism of MPTP by cytochrome P4502D6 and the demonstration of 2D6 mRNA in human foetal and adult brain by in situ hybridization. Xenobiotica 1997, 27, 111–125.
- Miksys, S.L.; Tyndale, R.F. Drug-metabolizing cytochrome P450s in the brain. J. Psychiatry Neurosci. 2002, 27, 406–415.
- McFadyen, M.C.; Melvin, W.T.; Murray, G.I. Cytochrome P450 in normal human brain and brain tumours. Biochem. Soc. Trans. 1997, 25, S577.
- Mann, A.; Miksys, S.; Lee, A.; Mash, D.C.; Tyndale, R.F. Induction of the drug metabolizing enzyme CYP2D in monkey brain by chronic nicotine treatment. Neuropharmacology 2008, 55, 1147–1155.
- Siegle, I.; Fritz, P.; Eckhardt, K.; Zanger, U.M.; Eichelbaum, M. Cellular localization and regional distribution of CYP2D6 mRNA and protein expression in human brain. Pharmacogenetics 2001, 11, 237–245.
- Dutheil, F.; Dauchy, S.; Diry, M.; Sazdovitch, V.; Cloarec, O.; Mellottée, L.; Bièche, I.; Ingelman-Sundberg, M.; Flinois, J.P.; de Waziers, I.; et al. Xenobiotic-metabolizing enzymes and transporters in the normal human brain: Regional and cellular mapping as a basis for putative roles in cerebral function. Drug Metab. Dispos. 2009, 37, 1528–1538.
- Hiroi, T.; Imaoka, S.; Funae, Y. Dopamine formation from tyramine by CYP2D. Biochem. Biophys. Res. Commun. 1998, 249, 838–843.
- Bromek, E.; Haduch, A.; Daniel, W.A. The ability of cytochrome P450 2D isoforms to synthesize dopamine in the brain: An in vitro study. Eur. J. Pharm. 2010, 626, 171–178.
- Yu, A.M.; Idle, J.R.; Byrd, L.G.; Krausz, K.W.; Küpfer, A.; Gonzalez, F.J. Regeneration of serotonin from 5-methoxytryptamine by polymorphic human CYP2D. Pharmacogenetics 2003, 13, 173–181.
- Yu, A.M.; Idle, J.R.; Gonzalez, F.J. Polymorphic cytochrome P450 2D6: Humanized mouse model and endogenous substrates. Drug Metab. Rev. 2004, 36, 243–277.
- Peñas-Lledó, E.M.; Llerena, A. CYP2D6 variation, behaviour and psychopathology: Implications for pharmacogenomics-guided clinical trials. Br. J. Clin. Pharm. 2014, 77, 673–683.
- Lalovic, B.; Phillips, B.; Risler, L.L.; Howald, W.; Shen, D.D. Quantitative Contribution of Cyp2d6 and Cyp3a to Oxycodone Metabolism in Human Liver and Intestinal Microsomes. Drug Metab. Dispos. 2004, 32, 447–454.
- Cone, E.J.; Darwin, W.D.; Buchwald, W.F.; Gorodetzky, C.W. Oxymorphone metabolism and urinary excretion in human, rat, guinea pig, rabbit, and dog. Drug Metab. Dispos. 1983, 11, 446–450.
- Rasmussen, I. Identification of Cytochrome P450 Isoforms Involved ín the Metabolísm oÍ Oxycodone. Master’s Thesis, Universify of Oslo, Oslo, Norway, December 2000.
- Romand, S.; Spaggiari, D.; Marsousi, N.; Samer, C.; Desmeules, J.; Daali, Y.; Rudaz, S. Characterization of oxycodone in vitro metabolism by human cytochromes P450 and UDP-glucuronosyltransferases. J. Pharm. Biomed. Anal. 2017, 144, 129–137.
- Fritz, A.; Busch, D.; Lapczuk, J.; Ostrowski, M.; Drozdzik, M.; Oswald, S. Expression of clinically relevant drug-metabolizing enzymes along the human intestine and their correlation to drug transporters and nuclear receptors: An intra-subject analysis. Basic Clin. Pharm. Toxicol. 2019, 124, 245–255.
- Pöyhiä, R.; Olkkola, K.T.; Seppälä, T.; Kalso, E. The pharmacokinetics of oxycodone after intravenous injection in adults. Br. J. Clin. Pharm. 1991, 32, 516–518.
- Korjamo, T.; Tolonen, A.; Ranta, V.-P.; Turpeinen, M.; Kokki, H. Metabolism of Oxycodone in Human Hepatocytes from Different Age Groups and Prediction of Hepatic Plasma Clearance. Front. Pharm. 2012, 2, 87.
- Pöyhiä, R.; Seppälä, T.; Olkkola, K.T.; Kalso, E. The pharmacokinetics and metabolism of oxycodone after intramuscular and oral administration to healthy subjects. Br. J. Clin. Pharm. 1992, 33, 617–621.
- Baselt, R.C.; Stewart, C.B. Determination of Oxycodone and a Major Metabolite in Urine by Electron-Capture GLC. J. Anal. Toxicol. 1978, 2, 107–109.
- Weinstein, S.H.; Gaylord, J.C. Determination of oxycodone in plasma and identification of a major metabolite. J. Pharm. Sci. 1979, 68, 527–528.
- Lalovic, B.; Kharasch, E.; Hoffer, C.; Risler, L.; Liu-Chen, L.-Y.; Shen, D.D. Pharmacokinetics and pharmacodynamics of oral oxycodone in healthy human subjects: Role of circulating active metabolites. Clin. Pharmacol. Ther. 2006, 79, 461–479.
- Nieminen, T.H.; Hagelberg, N.M.; Saari, T.I.; Pertovaara, A.; Neuvonen, M.; Laine, K.; Neuvonen, P.; Olkkola, K.T. Rifampin Greatly Reduces the Plasma Concentrations of Intravenous and Oral Oxycodone. Anesthesiology 2009, 110, 1371–1378.
- Sirhan-Daneau, A.M.V.; Manzini, C.; Schwab, R.; Demers, A.; Roy, I.; Lafrenchi, P.; Chauny, J.M.; St-Onge, M.; Gaudette, F.; Belanger, F.; et al. Role of CYP2D6 in the pharmacokinetics and pharmacodynamics of oxycodone in healthy volunteers during co-treatment with placebo or quinidine. Can. J. Clin. Pharm. 2010, 17, 1.
- Sirhan-Daneau, A.M.V.; St-Onge, M.; Turgeon, J. Pharmacokinetics of oxycodone in extensive and poor metabolizers of CYP2D6 during co-treatment with placebo or quinidine. In Proceedings of the European ISSX Meeting, Lisbon, Portugal, 17–20 May 2009.
- Samer, C.F.; Daali, Y.; Wagner, M.; Hopfgartner, G.; Eap, C.; Rebsamen, M.C.; Rossier, M.; Hochstrasser, D.; Dayer, P.; Desmeules, J.A. The effects of CYP2D6 and CYP3A activities on the pharmacokinetics of immediate release oxycodone. Br. J. Pharmacol. 2010, 160, 907–918.
- Andreassen, T.N.; Eftedal, I.; Klepstad, P.; Davies, A.; Bjordal, K.; Lundström, S.; Kaasa, S.; Dale, O. Do CYP2D6 genotypes reflect oxycodone requirements for cancer patients treated for cancer pain? A cross-sectional multicentre study. Eur. J. Clin. Pharmacol. 2011, 68, 55–64.
- Stamer, U.M.; Zhang, L.; Book, M.; Lehmann, L.E.; Stuber, F.; Musshoff, F. CYP2D6 Genotype Dependent Oxycodone Metabolism in Postoperative Patients. PLoS ONE 2013, 8, e60239.
- Kawakami, M.; Takenoshita-Nakaya, S.; Takeba, Y.; Nishimura, Y.; Oda, M.; Watanabe, M.; Ohta, Y.; Kobayashi, S.; Ohtsubo, T.; Kobayashi, S.; et al. Evaluation of CYP2D6 Protein Expression and Activity in the Small Intestine to Determine Its Metabolic Capability in the Japanese Population. Biol. Pharm. Bull. 2017, 40, 1344–1351.
- Paine, M.F.; Hart, H.L.; Ludington, S.S.; Haining, R.L.; Rettie, A.E.; Zeldin, D.C. The Human Intestinal Cytochrome P450 “Pie”. Drug Metab. Dispos. 2006, 34, 880–886.
- Li, A.P.; Ho, M.D.; Alam, N.; Mitchell, W.; Wong, S.; Yan, Z.; Kenny, J.R.; Hop, C.E.C.A. Inter-individual and inter-regional variations in enteric drug metabolizing enzyme activities: Results with cryopreserved human intestinal mucosal epithelia (CHIM) from the small intestines of 14 donors. Pharmacol. Res. Perspect. 2020, 8, e00645.
- Gaudette, F.; Sirhan-Daneau, A.; St-Onge, M.; Turgeon, J.; Michaud, V. Development of a sensitive method for the determination of oxycodone and its major metabolites noroxycodone and oxymorphone in human plasma by liquid chromatography–tandem mass spectrometry. J. Chromatogr. B 2016, 1008, 174–180.
- Heiskanen, T.; Olkkola, K.T.; Kalso, E. Effects of blocking CYP2D6 on the pharmacokinetics and pharmacodynamics of oxycodone*. Clin. Pharmacol. Ther. 1998, 64, 603–611.
- Lemberg, K.; Heiskanen, T.; Neuvonen, M.; Kontinen, V.; Neuvonen, P.; Dahl, M.-L.; Kalso, E. Does co-administration of paroxetine change oxycodone analgesia: An interaction study in chronic pain patients. Scand. J. Pain 2010, 1, 24–33.
- Hagelberg, N.M.; Nieminen, T.H.; Saari, T.I.; Neuvonen, M.; Neuvonen, P.J.; Laine, K.; Olkkola, K.T. Voriconazole drastically increases exposure to oral oxycodone. Eur. J. Clin. Pharmacol. 2008, 65, 263–271.
- Saari, T.I.; Grönlund, J.; Hagelberg, N.M.; Neuvonen, M.; Laine, K.; Neuvonen, P.; Olkkola, K.T. Effects of itraconazole on the pharmacokinetics and pharmacodynamics of intravenously and orally administered oxycodone. Eur. J. Clin. Pharmacol. 2010, 66, 387–397.
- Grönlund, J.; Saari, T.; Hagelberg, N.; Martikainen, I.K.; Neuvonen, P.J.; Olkkola, K.T.; Laine, K. Effect of Telithromycin on the Pharmacokinetics and Pharmacodynamics of Oral Oxycodone. J. Clin. Pharmacol. 2010, 50, 101–108.
- Boström, E.; Hammarlund-Udenaes, M.; Simonsson, U. Blood–Brain Barrier Transport Helps to Explain Discrepancies in In Vivo Potency between Oxycodone and Morphine. Anesthesiology 2008, 108, 495–505.
- Boström, E.; Simonsson, U.S.H.; Hammarlund-Udenaes, M. In Vivo Blood-Brain Barrier Transport of Oxycodone in the Rat: Indications for Active Influx and Implications for Pharmacokinetics/Pharmacodynamics. Drug Metab. Dispos. 2006, 34, 1624–1631.
- Okura, T.; Higuchi, K.; Deguchi, Y. The Blood-Brain Barrier Transport Mechanism Controlling Analgesic Effects of Opioid Drugs in CNS. Yakugaku Zasshi 2015, 135, 697–702.
- Sadiq, M.W.; Boström, E.; Keizer, R.; Bjorkman, S.; Hammarlund-Udenaes, M. Oxymorphone Active Uptake at the Blood–Brain Barrier and Population Modeling of its Pharmacokinetic–Pharmacodynamic Relationship. J. Pharm. Sci. 2013, 102, 3320–3331.
- Samer, C.F.; Daali, Y.; Wagner, M.; Hopfgartner, G.; Eap, C.; Rebsamen, M.C.; Rossier, M.; Hochstrasser, D.; Dayer, P.; Desmeules, J.A. Genetic polymorphisms and drug interactions modulating CYP2D6 and CYP3A activities have a major effect on oxycodone analgesic efficacy and safety. Br. J. Pharmacol. 2010, 160, 919–930.
- Coffman, B.L.; King, C.D.; Rios, G.R.; Tephly, T.R. The glucuronidation of opioids, other xenobiotics, and androgens by human UGT2B7Y(268) and UGT2B7H(268). Drug Metab. Dispos. 1998, 26, 73–77.
- Adams, M.P.; Ahdieh, H. Single- and Multiple-Dose Pharmacokinetic and Dose-Proportionality Study of Oxymorphone Immediate-Release Tablets. Drugs 2005, 6, 91–99.
- Adams, M.P.; Ahdieh, H. Pharmacokinetics and dose-proportionality of oxymorphone extended release and its metabolites: Results of a randomized crossover study. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2004, 24, 468–476.
This entry is offline, you can click here to edit this entry!