Crohn’s disease (CD) is a chronic disorder characterized by full thickness patchy inflammation of the gastrointestinal tract. The pathogenesis is multifactorial and involves defective innate immune responses, microbiome alterations, and dysregulated activation of the acquired component of mucosal immunity. One of the molecular mediators that is involved at different levels in the initiation and progression of intestinal inflammation characteristic of CD is tumor necrosis factor (TNF).
- Crohn’s disease
- tumor necrosis factor
- anti-TNF
- infliximab
- adalimumab
- certolizumab
- innate immunity
1. TNF in The Pathogenesis of Crohn’s Disease: A Dichotomous Role
TNF is a homotrimer of 157 amino acid subunits, primarily produced in soluble or transmembrane form by activated macrophages, although other immune cells, such as dendritic cells, T cells, B cells, and mast cells—but also epithelial and endothelial cells and fibroblasts—can be induced to express TNF [1]. TNF exerts its functions by the interaction with two membrane receptors, TNFR1 and TNFR2. The first is expressed by every type of cell, has a cytoplasmic death domain, and probably mediates most of the biologic effects of TNF. After binding of soluble or membrane-bound TNF, the inhibitory protein silencer of death domains (SODD) is released from TNFR1′s intracellular domain (ICD). It is then recognized by the adaptor protein TNF receptor–associated death domain (TRADD), which in turn recruits additional adaptor proteins such as receptor-interacting protein (RIP), TNFR-associated factor 2 (TRAF2), and Fas-associated death domain (FADD). Different pathways are activated, resulting in apoptosis induction by caspases upon the initial cleavage of caspase 8 and the activation of two major transcription factors such as c-Jun and NF-κB. These transcription factors regulate a large number of genes involved in proliferation, inflammation, and immune response. TNFR2 expression is limited to endothelial, hematopoietic, and immune cells, and after binding with TNF, it recruits TRAF2 and initiates a molecular cascade that finally results in kinase and NF-κB activation. Extensive crosstalk exists among the different pathways stimulated by TNF at the cellular level, and multiple biological effects may result in the stimulation of different cell types by TNF [2]. In addition, the pleiotropic effect on apoptosis regulation by TNF may be related to the crosstalk between this molecule and other receptors, such as receptor tyrosine kinases (RTKs) [3], or other pathways, such as the Ras-association domain family member (RASSF)-dependent activation of Hippo pathway [4].
1.1. The Classical Pro-Inflammatory Effect of TNF
TNF was identified in 1975 and purified and cloned in 1985 [5][6]. Initial research focused on the potential clinical applications associated with anti-neoplastic effects observed in pre-clinical studies. However, clinical trials subsequently showed disappointing results for lack of clinical benefits and high toxicity. Afterwards, the observation of increased TNF in serum and feces of IBD patients, and the characterization of experimental models of TNF-dependent systemic and intestinal inflammation opened the door to identifying the pro-inflammatory effects of TNF and its potential relevance in IBD pathogenesis [7]. During inflammation, TNF is a crucial mediator in the late phase of the inflammatory cascade. TNF is also responsible for the maintenance and chronicity of intestinal mucosal inflammation through recruitment and activation of lymphocytes and granulocytes, local expression of adhesion molecules on endothelial cells at the site of inflammation, secretion of pro-inflammatory mediators (IFNγ and IL-6) and radical oxygen species, and the formation of edema and granulomata [8]. The relevant pro-inflammatory role of TNF in IBD is definitively confirmed by the impact that the drugs blocking TNF have had on the management of IBD patients, not only for symptom relief, but for the effects on more consistent outcomes of disease such as mucosal healing and reduced surgery [9].
1.2. Potential Protective and Anti-Inflammatory Effect of TNF
Aside from its proinflammatory function, consistent data exist regarding the role of TNF in mucosal homeostasis. TNF represents a fundamental cytokine of innate immunity that has an important role in maintaining the homeostatic balance between luminal contents (i.e., microbiota, pathogenic bacteria) and the mucosal immune system, actively participating in intestinal barrier function. In an experimental model of intestinal inflammation, such as dextran sulfate sodium (DSS)-induced colitis, an incremental increase in colitis severity was observed in mice after monoclonal anti-TNF administration [10][11]. Additionally, immunodeficient mice (RAG knockout (KO)) that lacked expression of TNFR1 had higher mortality and impaired epithelial regeneration [12]. Similar results were observed in another model of experimental colitis (2,4,6-Trinitrobenzenesulfonic acid (TNBS)) [13]. The exact molecular pathways that mediate the protective effects of TNF on the intestinal mucosa are not completely clear, but several hypotheses can be considered. In particular, the different anti- vs. pro-inflammatory effects of TNF could be caused by different cellular sources and/or mucosal site/targets of TNF, different responses elicited, and the direct effects on enhancing intestinal barrier integrity. Most of the TNF involved in the inflammatory cascade is produced by activated macrophages and lymphocytes, whereas smaller amounts can be produced by epithelial cells, and that may be partly directed against luminal bacteria and partly stimulate the enhancement of barrier defense by means of reducing permeability and stimulating protective factors production, such as chemokines (i.e., defensins). In a spontaneous mouse model closely resembling human CD, the SAMP1/YitFc (SAMP) model of CD-like ileitis, administration of a multispecies compound of probiotics at a high dose before the onset of intestinal disease effectively prevented intestinal inflammation by stimulation of epithelial TNF and a consequent permeability decrease, and interestingly, the preventive effect was abrogated by the administration of recombinant anti-TNF [14]. This finding was further confirmed by in vitro experimentation indicating that pre-treatment of ilea from pre-inflamed SAMP mice with probiotic-conditioned media or TNF decreased ileal paracellular permeability by modulating tight junction proteins [15]. The relevance of TNF and other innate cytokines in regard to intestinal homeostasis has also been confirmed by the link between the pattern recognition receptors (PRR) pathway and production of such molecules, mediated by the MyD88 signaling adaptor protein. In line with these findings, mice deficient in MyD88 had more severe colitis and higher morbidity after DSS administration, due to the lack of upregulation of innate cytokines, including TNF and IL-1β [16]. Another mechanism by which TNF may exert its protective role within the intestinal mucosa is by limiting the progression of inflammation through the stimulation of apoptosis of effector immunocytes within the lamina propria that have important roles in eliminating intruding luminal bacteria [17]. Moreover, TNF has been shown to stimulate the production of mucosal glucocorticosteroids with anti-inflammatory activity at mucosal sites after acute injury [18]. Recently, a TNFR2-mediated regulatory effect of TNF on the expression of IL17 in Treg lymphocytes was shown, suggesting a potential reciprocal regulation between these two cytokines [19][20]. Although consistent crosstalk and overlapping functions exist between TNFR1 and TNFR2, the latter is likely to electively mediate homeostatic effects such as cell proliferation/survival and tissue regeneration [21].
1.3. Potential Clinical Implications
Taken together, those findings suggest a definite distinct role of TNF in the early vs. late phase of the inflammatory process leading to Crohn’s disease [22]. In the early phase, as a breach in the innate immune defense and of the intestinal barrier seems to be the relevant momentum for the initiation of the inflammatory cascade, TNF may contribute to straighten the innate response in that crucial phase. In the late stage, TNF substantially contribute to the maintenance of the chronic inflammation characteristic of the active state of the disease (Figure 1). Drugs blocking TNF in that phase of the disease may consistently reduce the inflammatory state, with evident clinical short- and long-term benefits that will be discussed in the next section. Nonetheless, since many patients do not respond to anti-TNF therapy, it could be possible that in those patients, the inhibition of protective role of TNF prevails over the anti-inflammatory effect consequent to its block. Another potential implication of the dual role of anti-TNF in the different disease phases regards the appropriate timing of the pharmacological block of the TNF. Early intervention is generally associated with a higher response rate, but the positive pre-clinical role of the TNF in the maintenance of mucosal homeostasis may be considered for the future design of clinical trials, considering with high caution very early or even preventive treatments involving TNF block. Indeed, the growing clinical data (which will be further discussed in the next section) indicating a better clinical efficacy of anti-TNF in early vs. long-standing CD mainly refers to the time from the diagnosis of the disease, which probably already corresponds to the late phase of the pathogenetic inflammatory process. Conversely, the early inflammatory phase, where TNF may exert a protective role, is essentially pre-clinical, whereas symptoms (that may lead to the diagnosis) occur only when the inflammatory process is fully established. In patients with long-lasting CD, the inflammatory burden tends to be less prominent, while fibro-stenotic complications increase. Notwithstanding the pro-fibrotic effect of TNF [23], a potential preventive role of anti-TNF drugs on strictures in CD patients is not demonstrated, so that a more consistent effect of that drugs is observed in early uncomplicated disease. Moreover, since pro-inflammatory cytokines’ cascade includes many overlapping and interacting pathways, TNF appears to have a regulatory role for other pro-inflammatory mediators that may be overstimulated after its block. Therefore, in particular for complicated patients, the increased comprehension of such molecular pathways may lead to the development of future therapeutic strategies implying multiple simultaneous pro-inflammatory pathways block.
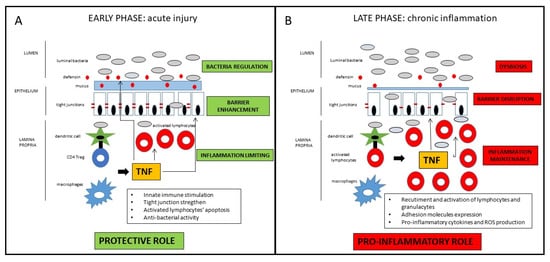
Figure 1. Schematic representation of the dichotomous role of TNF in CD pathogenesis. In the early phase of the inflammatory process, TNF has protective effects on the intestinal mucosa, whereas in the late phase of disease, TNF has consistent pro-inflammatory effects. For detailed description, please refer to the text.
2. The TNF Block as a Therapeutic Target in Crohn’s Disease: Current Drugs Available
There are currently three anti-TNF drugs approved by the US Food and Drug Administration (FDA) for the treatment of CD patients: infliximab, adalimumab, and certolizumab pegol. Certolizumab pegol is not approved by European regulatory agency (European Medicine Agency—EMA) [24].
Infliximab is a chimeric IgG1 monoclonal antibody against TNF, and it was the first anti-TNF drug approved by FDA for the treatment of CD (in 1998, and by EMA in 1999), first demonstrating efficacy in 1997 [25]. It can fix, complement, and lyse cells expressing membrane-bound anti-TNF with a potent anti-inflammatory effect at the intestinal mucosal site involving probably more unknown pathways. It is administered in intravenous infusions at weeks 0, 2, and 6 (induction) and then every 8 weeks (maintenance). Pivotal trials demonstrating its efficacy in CD patients are the ACCENT I [26] and II [27], as well as the SONIC trial [28], but more trials and real-life studies further confirmed drug efficacy and safety.
Adalimumab is a human IgG1 anti-TNF antibody that was approved for clinical use in CD patients by the FDA in 2002 and by the EMA in 2003. It has the ability to fix complement and lyse cell expressing TNF, and it is administered by subcutaneous injection every other week. Among many studies demonstrating efficacy and safety of that drug in CD patients, the CLASSIC I and II [29][30] trials demonstrated efficacy in induction and maintenance of remission, while CHARM [31], GAIN [32], and Extend [33] trials proved the efficacy of the drug in fistulizing disease, as a second line after infliximab failure, and for mucosal healing achievement.
Certolizumab pegol is a PEGylated Fab’ fragment of a humanized monoclonal anti-TNF antibody with a high binding affinity for soluble and transmembrane TNF and less immunogenicity side effects due to the lack of the Fc portion. It does not induce apoptosis nor fix complement. It is administered subcutaneously and, having a longer half-life than adalimumab due to the PEG addition, has a maintenance dosing every month. The registrative trials PRECISE 1 [34], PRECISE 2 [35], and PRECISE 3 [36] demonstrated the efficacy of the drug in CD patients, but relevant endpoints, such as induction of remission and response rate over placebo at week 6 and 26, were not achieved. The drug is not approved for clinical utilization for CD by the EMA, but it was approved by the FDA in 2008 for the treatment of signs and symptoms in CD patients who fail to respond to other conventional treatments.
Data of the main clinical trials of the three anti-TNF agents currently used in CD patients are summarized in Table 1. Those drugs, which so profoundly revolutionized the therapeutic approach in IBD patients, still represent the cornerstone of treatment for moderate–severe CD. The efficacy of anti-TNF in inducing and maintaining of response and remission in CD patients has been further confirmed by a network meta-analysis including 10 studies, with no clear superiority among drugs [37]. Indeed, besides the efficacy for inflammatory luminal disease, such therapies have been proven beneficial in specific complex clinical situations, such as prevention of post-operative recurrence [38], peri-anal fistulizing disease [39], concurrent extra-intestinal manifestation [40], and small bowel stenosis [41]. Therefore, notwithstanding the novel drugs already available, anti-TNF agents (infliximab, adalimumab, and certolizumab) still represent indispensable tools in the therapeutic armamentarium for CD management.
Table 1. Pivotal clinical trials of the three anti-TNF drugs approved for CD patients.
| Drug | Trials (Ref) | Patients | End-Points | Results |
|---|---|---|---|---|
| Infliximab (IFX) | ACCENT I (72) ACCENT II (73) SONIC (74) |
335/573 responders at week 2 282 222 |
Remission at week 30 Fistulas’ healing at week 54 Steroid-free remission and mucosal healing at week 26 |
IFX 5 mg/Kg: 44/113 (39%) IFX 10 mg/Kg: 50/112 (45%) Placebo: 23/110 (21%) IFX induction + 8 week maintenance: 50/138 (36%) IFX induction + placebo: 27/144 (19%) Remission—IFX + AZA: 96/169 (57%) IFX: 75/169 (44%) AZA: 51/170 (30%) Mucosal healing—IFX + AZA: 47/107 (44%) IFX: 28/93 (30%) AZA 18/109 (17%) |
| Adalimumab (ADA) | CLASSIC I (75) CLASSIC II(76) CHARM (77) GAIN (78) Extend (79) |
299 259 499/854 responders at week 4 325 135 |
Remission at week 4 Remission at week 56 Remission at week 26 and 56 Fistulas healing at week 56 Remission rate at week 4 in patients IFX non responders Mucosal healing at week 12 and 52 |
ADA 160/80 mg: 27/76 (36%) ADA 80/40 mg: 18/75 (24%) ADA 40/20 mg: 13/74 (18%) Placebo: 9/74 (12%) ADA 40 mg/2 week: 15/19 (79%) ADA 40 mg/week: 15/18 (83%) Placebo: 8/18 (44%) ADA with dose optimization: 93/204 (46%) Week 26—ADA 40 mg/2 week: 68/172 (40%) ADA 40 mg/week: 75/157 (47%) Placebo: 29/170 (17%) Week 56—ADA 40 mg/2 week: 62/172 (36%) ADA 40 mg/week: 65/157 (41%) Placebo: 20/170 (12%) ADA 40 mg/2 week: 10/30 (33%) ADA 40 mg/week: 11/40 (28%) Placebo: 6/47 (13%) ADA: 34/159 (21%) Placebo: 12/166 (7%) Week 12—ADA 40 mg/2 week: 17/62 (27%) Placebo: 8/61 (13%) Week 52—ADA 40 mg/2 week: 15/62 (24%) Placebo: 0/61 (0%) |
| Certolizumab pegol (CZP) | PRECISE1 (80) PRECISE2 (81) PRECISE3 (82) |
655 425 241 from PRECISE2 |
Remission at week 6 and 26 Remission at week 26 Remission at week 52 and 80 |
Week 6—CZP: 71/329 (22%) Placebo: 57/326 (17%) Week 6 and 26—CZP: 47/327 (14%) Placebo: 32/326 (10%) CZP: 103/215 (48%) Placebo: 61/210 (29%) Week 52—CZP continuous group: 58/141 (41%) Drug-interruption group *: 30/100 (30%) Week 80—CZP continuous group: 51/141 (36%) Drug-interruption group *: 23/100 (23%) |
* Patients who had CZP from week 0 to 6, then placebo from week 6 to 26 (from PRECISE2), then CZP to week 80 (in PRECISE3).
Other anti-TNF drugs did not show efficacy in RCTs in CD patients including: etanercept [42], a soluble form of the p75 receptor that inhibits TNF, already approved for rheumatologic and dermatologic indications; CDP571 [43], an engineered human monoclonal antibody against TNF; and onercept [44], a recombinant soluble human p55 receptor to TNF. The reasons for the lack of efficacy of such anti-TNF agents are not completely clear: CDP571 is an IgG4 antibody, while infliximab and adalimumab are IgG1, and such a difference may lead to some alterations that may be relevant for the clinical actions. For etanercept and onercept, the crucial issue is probably the fact that they bind only the soluble form of TNF, which is most likely not sufficient to determine clinical effects in CD. Moreover, since etanercept is largely used in rheumatology and dermatology, consistent risk for new onset or exacerbation of IBD has been described [45][46]. A possible explanation is that, as etanercept is a soluble TNFRII receptor, it binds not only the TNF but even other ligands such as lymphotoxin-α, which is a relevant cytokine for mucosal homeostasis [47]. Besides etanercept, the possible onset of IBD in patients treated with others anti-TNF for non-gastroenterological indications is rarely reported [45], further reflecting the complexity and the redundancy of the TNF pathway, and the possibility that, in a subset of patients, the block of TNF may worsen the inflammatory process.
Despite the widespread use of anti-TNF agents since the last two decades, some open issues are still up for debate. According to clinical trials and real-life studies, about 20% of patients do not respond to anti-TNF ab initio (primary failure), and about 20–30% of patients are likely to lose response every year (secondary failure). Therefore, a consistent portion of patients would need a second-line therapy or a surgical approach. In order to increase efficacy of therapy and to ideally affect the natural course of the disease, “early treatment” has been proposed—by 2 years from the initial diagnosis—in selected CD patients with features of severe disease (i.e., young age, extensive intestinal involvement, perianal disease, smokers). In the SUTD (Step Up vs. Top Down) trial, an early immunosuppression plus infliximab administration was associated with a higher clinical remission rate at 2 years in 133 CD patients compared to patients with conventional management [48]. In the REACT trial, in a larger set of patients (more than 1700) comparing early immunosuppression plus adalimumab vs. conventional management, only a trend for a higher rate of clinical remission at 2 years was found in the early treatment group, but when more consistent outcomes were considered (hospital admission, surgery, and serious disease-related complications), a significant difference emerged [49].
The observation that anti-drug antibodies are associated with infusion reactions and loss of response—and that therapeutic efficacy is coupled with adequate blood drug level—has pushed toward the concept that the measurement of such parameters (i.e., anti-TNF Ab and drug trough levels) would optimize anti-TNF drug utilization [50]. Therapeutic drug monitoring (TDM) can be used in two main clinical settings: in primary and secondary biologic failure patients in order to more appropriately assess consequent therapeutic strategy such as optimization, switch-in-class, switch-out-of-class (reactive TDM) or in patients in remission to drive to a tailored approach to drug administration to maintain efficacy and minimize side-effects (pro-active TDM) [51]. Despite the rational and the cost-saving effect, no decisive evidence is available for a significant superiority of TDM over the sole clinical approach both for the reactive and pro-active monitoring. Yanai et al. demonstrated in a retrospective study that TDM can help in the management of patients who lost response to infliximab and adalimumab [52]. A Danish cohort study indicated that TDM-guided management had a similar clinical outcome at 12 weeks as an empirical approach, but was cost-effective [53], and some studies confirmed that finding [54][55]. For pro-active TDM, even less evidence is available, and the two main trials addressing the potential utility of that approach; i.e., the TAXIT and TAILORIX trials failed to show significant benefits over the clinical based management [56][57]. Considering that—and the fact that controversies exist for assay methods and reference levels—TDM is generally not routinely performed in clinical practice, but it can be useful in the management of anti-TNF failure patients.
This entry is adapted from the peer-reviewed paper 10.3390/ijms221910273
References
- Chen, G.; Goeddel, D.V. TNF-R1 signaling: A beautiful pathway. Science 2002, 296, 1634–1635.
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7, 91.
- Beyer, E.M.; MacBeath, G. Cross-talk between receptor tyrosine kinase and tumor necrosis factor-alpha signaling networks regulates apoptosis but not proliferation. Mol. Cell. Proteom. MCP 2012, 11, M111.013292.
- Oceandy, D.; Amanda, B.; Ashari, F.Y.; Faizah, Z.; Azis, M.A.; Stafford, N. The Cross-Talk Between the TNF-alpha and RASSF-Hippo Signalling Pathways. Int. J. Mol. Sci. 2019, 20, 2346.
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666–3670.
- Aggarwal, B.B.; Kohr, W.J. Human tumor necrosis factor. Methods Enzymol. 1985, 116, 448–456.
- D’Haens, G.R.; van Deventer, S. 25 years of anti-TNF treatment for inflammatory bowel disease: Lessons from the past and a look to the future. Gut 2021, 70, 1396–1405.
- Papadakis, K.A.; Targan, S.R. Tumor necrosis factor: Biology and therapeutic inhibitors. Gastroenterology 2000, 119, 1148–1157.
- Van Assche, G.; Vermeire, S.; Rutgeerts, P. The potential for disease modification in Crohn’s disease. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 79–85.
- Kojouharoff, G.; Hans, W.; Obermeier, F.; Mannel, D.N.; Andus, T.; Scholmerich, J.; Gross, V.; Falk, W. Neutralization of tumour necrosis factor (TNF) but not of IL-1 reduces inflammation in chronic dextran sulphate sodium-induced colitis in mice. Clin. Exp. Immunol. 1997, 107, 353–358.
- Naito, Y.; Takagi, T.; Handa, O.; Ishikawa, T.; Nakagawa, S.; Yamaguchi, T.; Yoshida, N.; Minami, M.; Kita, M.; Imanishi, J.; et al. Enhanced intestinal inflammation induced by dextran sulfate sodium in tumor necrosis factor-alpha deficient mice. J. Gastroenterol. Hepatol. 2003, 18, 560–569.
- Mizoguchi, E.; Hachiya, Y.; Kawada, M.; Nagatani, K.; Ogawa, A.; Sugimoto, K.; Mizoguchi, A.; Podolsky, D.K. TNF receptor type I-dependent activation of innate responses to reduce intestinal damage-associated mortality. Gastroenterology 2008, 134, 470–480.
- Ebach, D.R.; Newberry, R.; Stenson, W.F. Differential role of tumor necrosis factor receptors in TNBS colitis. Inflamm. Bowel Dis. 2005, 11, 533–540.
- Pagnini, C.; Saeed, R.; Bamias, G.; Arseneau, K.O.; Pizarro, T.T.; Cominelli, F. Probiotics promote gut health through stimulation of epithelial innate immunity. Proc. Natl. Acad. Sci. USA 2010, 107, 454–459.
- Corridoni, D.; Pastorelli, L.; Mattioli, B.; Locovei, S.; Ishikawa, D.; Arseneau, K.O.; Chieppa, M.; Cominelli, F.; Pizarro, T.T. Probiotic bacteria regulate intestinal epithelial permeability in experimental ileitis by a TNF-dependent mechanism. PLoS ONE 2012, 7, e42067.
- Rakoff-Nahoum, S.; Paglino, J.; Eslami-Varzaneh, F.; Edberg, S.; Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004, 118, 229–241.
- Atreya, R.; Zimmer, M.; Bartsch, B.; Waldner, M.J.; Atreya, I.; Neumann, H.; Hildner, K.; Hoffman, A.; Kiesslich, R.; Rink, A.D.; et al. Antibodies against tumor necrosis factor (TNF) induce T-cell apoptosis in patients with inflammatory bowel diseases via TNF receptor 2 and intestinal CD14(+) macrophages. Gastroenterology 2011, 141, 2026–2038.
- Noti, M.; Corazza, N.; Mueller, C.; Berger, B.; Brunner, T. TNF suppresses acute intestinal inflammation by inducing local glucocorticoid synthesis. J. Exp. Med. 2010, 207, 1057–1066.
- Urbano, P.C.M.; He, X.; van Heeswijk, B.; Filho, O.P.S.; Tijssen, H.; Smeets, R.L.; Joosten, I.; Koenen, H. TNFalpha-Signaling Modulates the Kinase Activity of Human Effector Treg and Regulates IL-17A Expression. Front. Immunol. 2019, 10, 3047.
- Bystrom, J.; Clanchy, F.I.; Taher, T.E.; Mangat, P.; Jawad, A.S.; Williams, R.O.; Mageed, R.A. TNFalpha in the regulation of Treg and Th17 cells in rheumatoid arthritis and other autoimmune inflammatory diseases. Cytokine 2018, 101, 4–13.
- Tseng, W.-Y.; Huang, Y.-S.; Lin, H.-H.; Luo, S.-F.; McCann, F.; McNamee, K.; Clanchy, F.; Williams, R. TNFR signalling and its clinical implications. Cytokine 2018, 101, 19–25.
- Bamias, G.; Corridoni, D.; Pizarro, T.T.; Cominelli, F. New insights into the dichotomous role of innate cytokines in gut homeostasis and inflammation. Cytokine 2012, 59, 451–459.
- Curciarello, R.; Docena, G.H.; MacDonald, T.T. The Role of Cytokines in the Fibrotic Responses in Crohn’s Disease. Front. Med. 2017, 4, 126.
- Adegbola, S.O.; Sahnan, K.; Warusavitarne, J.; Hart, A.; Tozer, P. Anti-TNF Therapy in Crohn’s Disease. Int. J. Mol. Sci. 2018, 19, 2244.
- Targan, S.R.; Hanauer, S.B.; van Deventer, S.J.; Mayer, L.; Present, D.H.; Braakman, T.; DeWoody, K.L.; Schaible, T.F.; Rutgeerts, P.J. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group. N. Engl. J. Med. 1997, 337, 1029–1035.
- Hanauer, S.B.; Feagan, B.G.; Lichtenstein, G.R.; Mayer, L.F.; Schreiber, S.; Colombel, J.F.; Rachmilewitz, D.; Wolf, D.C.; Olson, A.; Bao, W.; et al. Maintenance infliximab for Crohn’s disease: The ACCENT I randomised trial. Lancet 2002, 359, 1541–1549.
- Sands, B.E.; Anderson, F.H.; Bernstein, C.N.; Chey, W.Y.; Feagan, B.G.; Fedorak, R.N.; Kamm, M.A.; Korzenik, J.R.; Lashner, B.A.; Onken, J.E.; et al. Infliximab maintenance therapy for fistulizing Crohn’s disease. N. Engl. J. Med. 2004, 350, 876–885.
- Colombel, J.F.; Sandborn, W.J.; Reinisch, W.; Mantzaris, G.J.; Kornbluth, A.; Rachmilewitz, D.; Lichtiger, S.; D’Haens, G.; Diamond, R.H.; Broussard, D.L.; et al. Infliximab, azathioprine, or combination therapy for Crohn’s disease. N. Engl. J. Med. 2010, 362, 1383–1395.
- Hanauer, S.B.; Sandborn, W.J.; Rutgeerts, P.; Fedorak, R.N.; Lukas, M.; MacIntosh, D.; Panaccione, R.; Wolf, D.; Pollack, P. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn’s disease: The CLASSIC-I trial. Gastroenterology 2006, 130, 323–333.
- Sandborn, W.J.; Hanauer, S.B.; Rutgeerts, P.; Fedorak, R.N.; Lukas, M.; MacIntosh, D.G.; Panaccione, R.; Wolf, D.; Kent, J.D.; Bittle, B.; et al. Adalimumab for maintenance treatment of Crohn’s disease: Results of the CLASSIC II trial. Gut 2007, 56, 1232–1239.
- Colombel, J.F.; Sandborn, W.J.; Rutgeerts, P.; Enns, R.; Hanauer, S.B.; Panaccione, R.; Schreiber, S.; Byczkowski, D.; Li, J.; Kent, J.D.; et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn’s disease: The CHARM trial. Gastroenterology 2007, 132, 52–65.
- Sandborn, W.J.; Rutgeerts, P.; Enns, R.; Hanauer, S.B.; Colombel, J.F.; Panaccione, R.; D’Haens, G.; Li, J.; Rosenfeld, M.R.; Kent, J.D.; et al. Adalimumab induction therapy for Crohn disease previously treated with infliximab: A randomized trial. Ann. Intern. Med. 2007, 146, 829–838.
- Rutgeerts, P.; Van Assche, G.; Sandborn, W.J.; Wolf, D.C.; Geboes, K.; Colombel, J.F.; Reinisch, W.; Investigators, E.; Kumar, A.; Lazar, A.; et al. Adalimumab induces and maintains mucosal healing in patients with Crohn’s disease: Data from the EXTEND trial. Gastroenterology 2012, 142, 1102–1111.e2.
- Sandborn, W.J.; Feagan, B.G.; Stoinov, S.; Honiball, P.J.; Rutgeerts, P.; Mason, D.; Bloomfield, R.; Schreiber, S.; Investigators, P.S. Certolizumab pegol for the treatment of Crohn’s disease. N. Engl. J. Med. 2007, 357, 228–238.
- Schreiber, S.; Khaliq-Kareemi, M.; Lawrance, I.C.; Thomsen, O.O.; Hanauer, S.B.; McColm, J.; Bloomfield, R.; Sandborn, W.J.; Investigators, P.S. Maintenance therapy with certolizumab pegol for Crohn’s disease. N. Engl. J. Med. 2007, 357, 239–250.
- Lichtenstein, G.R.; Thomsen, O.O.; Schreiber, S.; Lawrance, I.C.; Hanauer, S.B.; Bloomfield, R.; Sandborn, W.J.; Precise 3 Study, I. Continuous therapy with certolizumab pegol maintains remission of patients with Crohn’s disease for up to 18 months. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2010, 8, 600–609.
- Stidham, R.W.; Lee, T.C.; Higgins, P.D.; Deshpande, A.R.; Sussman, D.A.; Singal, A.G.; Elmunzer, B.J.; Saini, S.D.; Vijan, S.; Waljee, A.K. Systematic review with network meta-analysis: The efficacy of anti-TNF agents for the treatment of Crohn’s disease. Aliment. Pharmacol. Ther. 2014, 39, 1349–1362.
- Bakouny, Z.; Yared, F.; El Rassy, E.; Jabbour, R.; Hallit, R.; Khoury, N.; Honein, K.; Bou Jaoude, J. Comparative Efficacy of Anti-TNF Therapies for The Prevention of Postoperative Recurrence of Crohn’s Disease: A Systematic Review and Network Meta-Analysis of Prospective Trials. J. Clin. Gastroenterol. 2019, 53, 409–417.
- Panes, J.; Rimola, J. Perianal fistulizing Crohn’s disease: Pathogenesis, diagnosis and therapy. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 652–664.
- Peyrin-Biroulet, L.; Van Assche, G.; Gomez-Ulloa, D.; Garcia-Alvarez, L.; Lara, N.; Black, C.M.; Kachroo, S. Systematic Review of Tumor Necrosis Factor Antagonists in Extraintestinal Manifestations in Inflammatory Bowel Disease. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2017, 15, 25–36.e27.
- Rodriguez-Lago, I.; Hoyo, J.D.; Perez-Girbes, A.; Garrido-Marin, A.; Casanova, M.J.; Chaparro, M.; Fernandez-Clotet, A.; Castro-Poceiro, J.; Garcia, M.J.; Sanchez, S.; et al. Early treatment with anti-tumor necrosis factor agents improves long-term effectiveness in symptomatic stricturing Crohn’s disease. United Eur. Gastroenterol. J. 2020, 8, 1056–1066.
- Sandborn, W.J.; Hanauer, S.B.; Katz, S.; Safdi, M.; Wolf, D.G.; Baerg, R.D.; Tremaine, W.J.; Johnson, T.; Diehl, N.N.; Zinsmeister, A.R. Etanercept for active Crohn’s disease: A randomized, double-blind, placebo-controlled trial. Gastroenterology 2001, 121, 1088–1094.
- Sandborn, W.J.; Feagan, B.G.; Hanauer, S.B.; Present, D.H.; Sutherland, L.R.; Kamm, M.A.; Wolf, D.C.; Baker, J.P.; Hawkey, C.; Archambault, A.; et al. An engineered human antibody to TNF (CDP571) for active Crohn’s disease: A randomized double-blind placebo-controlled trial. Gastroenterology 2001, 120, 1330–1338.
- Rutgeerts, P.; Sandborn, W.J.; Fedorak, R.N.; Rachmilewitz, D.; Tarabar, D.; Gibson, P.; Haagen Nielsen, O.; Wild, G.; Schreiber, S.; Pena Rossi, C.; et al. Onercept for moderate-to-severe Crohn’s disease: A randomized, double-blind, placebo-controlled trial. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2006, 4, 888–893.
- Korzenik, J.; Larsen, M.D.; Nielsen, J.; Kjeldsen, J.; Norgard, B.M. Increased risk of developing Crohn’s disease or ulcerative colitis in 17 018 patients while under treatment with anti-TNFalpha agents, particularly etanercept, for autoimmune diseases other than inflammatory bowel disease. Aliment. Pharmacol. Ther. 2019, 50, 289–294.
- O’Toole, A.; Lucci, M.; Korzenik, J. Inflammatory Bowel Disease Provoked by Etanercept: Report of 443 Possible Cases Combined from an IBD Referral Center and the FDA. Dig. Dis. Sci. 2016, 61, 1772–1774.
- Levin, A.D.; Wildenberg, M.E.; Van den Brink, G.R. Mechanism of Action of Anti-TNF Therapy in Inflammatory Bowel Disease. J. Crohn’s Colitis 2016, 10, 989–997.
- D’Haens, G.; Baert, F.; van Assche, G.; Caenepeel, P.; Vergauwe, P.; Tuynman, H.; De Vos, M.; van Deventer, S.; Stitt, L.; Donner, A.; et al. Early combined immunosuppression or conventional management in patients with newly diagnosed Crohn’s disease: An open randomised trial. Lancet 2008, 371, 660–667.
- Khanna, R.; Bressler, B.; Levesque, B.G.; Zou, G.; Stitt, L.W.; Greenberg, G.R.; Panaccione, R.; Bitton, A.; Pare, P.; Vermeire, S.; et al. Early combined immunosuppression for the management of Crohn’s disease (REACT): A cluster randomised controlled trial. Lancet 2015, 386, 1825–1834.
- Mould, D.R.; Green, B. Pharmacokinetics and pharmacodynamics of monoclonal antibodies: Concepts and lessons for drug development. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2010, 24, 23–39.
- Mitrev, N.; Leong, R.W. Therapeutic drug monitoring of anti-tumour necrosis factor-alpha agents in inflammatory bowel disease. Expert Opin. Drug Saf. 2017, 16, 303–317.
- Yanai, H.; Lichtenstein, L.; Assa, A.; Mazor, Y.; Weiss, B.; Levine, A.; Ron, Y.; Kopylov, U.; Bujanover, Y.; Rosenbach, Y.; et al. Levels of drug and antidrug antibodies are associated with outcome of interventions after loss of response to infliximab or adalimumab. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2015, 13, 522–530.e2.
- Steenholdt, C.; Brynskov, J.; Thomsen, O.O.; Munck, L.K.; Fallingborg, J.; Christensen, L.A.; Pedersen, G.; Kjeldsen, J.; Jacobsen, B.A.; Oxholm, A.S.; et al. Individualised therapy is more cost-effective than dose intensification in patients with Crohn’s disease who lose response to anti-TNF treatment: A randomised, controlled trial. Gut 2014, 63, 919–927.
- Velayos, F.S.; Kahn, J.G.; Sandborn, W.J.; Feagan, B.G. A test-based strategy is more cost effective than empiric dose escalation for patients with Crohn’s disease who lose responsiveness to infliximab. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2013, 11, 654–666.
- Stein, B.N.; Pellish, R.S.; Thompson, K.D.; Baptista, V.; Siegel, C.A. Using Therapeutic Drug Monitoring to Identify Variable Infliximab Metabolism in an Individual Patient with Ulcerative Colitis. J. Clin. Gastroenterol. 2016, 50, 66–68.
- Vande Casteele, N.; Ferrante, M.; Van Assche, G.; Ballet, V.; Compernolle, G.; Van Steen, K.; Simoens, S.; Rutgeerts, P.; Gils, A.; Vermeire, S. Trough concentrations of infliximab guide dosing for patients with inflammatory bowel disease. Gastroenterology 2015, 148, 1320–1329.e3.
- D’Haens, G.R.; Vermeire, S.; Lambrecht, G.; Baert, F.J.; Bossuyt, P.; Nachury, M.; Buisson, A.; Bouhnik, Y.; Filippi, J.; Van der Woude, C.J. 692 Drug-level based dosing versus symptom-based dose adaptation in patients with Crohn’s disease: A prospective, randomized multicenter study (TAILORIX). Gastroenterology 2016, 150, S143.