Pancreatic Ductal Adenocarcinoma (PDAC) is an expeditiously fatal malignancy with a five-year survival rate of 6–8%. Conventional chemotherapeutics fail in many cases due to inadequate primary response and rapidly developing resistance. This treatment failure is particularly challenging in pancreatic cancer because of the high molecular heterogeneity across tumors. Additionally, a rich fibro-inflammatory component within the tumor microenvironment (TME) limits the delivery and effectiveness of anticancer drugs, further contributing to the lack of response or developing resistance to conventional approaches in this cancer. Patient-derived three-dimensional (3D) organoid technology has provided a unique opportunity to study patient-specific cancerous epithelium. Patient-derived organoids cultured with the TME components can more accurately reflect the in vivo tumor environment. A number of in vitro models have been developed to address the limitation of the lack of tumor extracellular matrix (ECM) in the conventional models of cancer and drug screening platforms. In this regard, microfluidic chips are cutting-edge devices that process fluids in micro-sized channels and allow the culture of multiple cell types within a matrix—so-called ‘organ-on-a-chip (OOC)’ technology. OOC allows us to recapitulate 3D multicellular architecture and microengineering of TME with the potential to bridge the gaps between bench and bedside by providing screening platforms for testing anticancer agents before reaching human clinical trials.
- pancreatic ductal adenocarcinoma
- microfluidics
- organ-on-a-chip
- tumor microenvironment
- tissue engineering
1. Organ-on-a-Chip (OOC) Technology in Cancer
Microfluidic chip devices are preferably fabricated on transparent surfaces such as glass or transparent polymer to make them amenable for microscopic imaging [1][2]. It is desirable for chip devices to be disposable. This makes the use of polymers attractive because of their safe and eco-friendly characteristics [3][4]. Polydimethylsiloxane (PDMS) has gained widespread adoption in the fields of the microfluidic chip, tissue engineering, and cancer biology due to its biocompatibility and ease of fabrication [2][5]. PDMS is oxygen permeable, supporting the culture of cells involved in PDAC TME, thus appropriate for cancer studies [6][7]. A detailed technical review of the materials and design of microfluidic devices is out of the scope of this article and can be found elsewhere [1][2][4][8].
In contrast to static cell culture systems in culture flasks and plates, a chip enables the modeling of in vivo physical conditions by allowing a controlled flow of culture medium into the cell chambers. The flow rates can be tailored to mimic the shear stress of the respective organ [8][9]. Our ability to mimic such dynamic cues (i.e., mechanical forces, hypoxia, and matrix stiffness) on a chip is crucial in modeling cellular events in cancer. For example, epithelial to mesenchymal transition (EMT), a key event in cancer progression and invasion, responds to such dynamic forces in the tumor [10][11][12][13]. The phenotypic transition of the cancerous epithelium into mesenchymal faith enhances their pro-survival tone, migratory capacity, and metastasis [12][14]. Dynamic laminar microfluidic platforms could show how flow-based shear stress promotes EMT in cancer [15][16]. Lung tumoroids culture in such flow-based microfluidic chips express higher EMT markers compared with tumoroids in static conditions [10]. Thus, the maneuverable features of microfluidic platforms allow us to evaluate the impact of physical stressors such as changes in the flow and shear stress on the mechanism of cancer progression.
The culture of several cell types within such a dynamic system has made microfluidic chips a desirable in vitro platform towards building more complex organ mimics [5][17][18]. Integration of additional devices and biosensors in the platform has further advanced its translational application for on-chip analysis. For instance, dielectrophoresis-type devices or dynamic-ELISA on a chip allow the measurement of secreted proteins and molecules in real-time [17][18][19][20][21][22]. Together, microfluidic devices incorporating multiple cell types in a physiologically relevant microenvironment with physical, biochemical, and optical sensing capabilities could be instrumental in better modeling molecular and/or cellular characterizations of cancer biology towards an individualized OOC platform for ex vivo drug testing in cancer [23]. This is important, as the TME, including vasculature, immune cells, and non-cellular components surrounding the cancerous epithelium, play a major role in tumor growth and drug resistance [24]. Here we will briefly list some of the crucial cellular processes occurring in the TME that have been successfully recapitulated in microfluidic devices.
1.1. Interaction of Cancerous Epithelium with Cellular Components of Tumor Microenvironment
When modeling a specific tumor on a chip, a desired goal is to include cancer cells among the other major cell types typically present in that TME [25]. Compared to traditional well-plate inserts, microfluidic channels provide adequate spatial organization and compartmentalization for culturing tumor spheroids and organoids with other cell types in vitro, where the interaction of the individual’s primary cancer cells could be studied with other components of the TME [26]. The co-culture of 3D tumor spheroids with cancer-associated fibroblasts (CAFs) within hydrogel scaffold on-chip has shown to be useful in studying cell–cell interactions [27]. In this study, the growth of human colorectal carcinoma cell spheroids was increased when co-cultured with fibroblasts. The co-culture enhanced fibroblast activation and migration, suggested bidirectional crosstalk between the cancer cells and the fibroblasts in the TME. When metastatic breast cancer cells were cultured with tumor-associated macrophages within a microfluidic channel, tumor-associated macrophages invaded areas containing the cancer cells [28]. In a pre-cancerous OOC model, co-culture of mammary epithelial cells with human mammary fibroblasts promoted normal ductal carcinoma transition to an invasive phenotype [29].
1.2. Angiogenesis
Tumor growth and metastasis are dependent on the formation of new blood vessels for vascular support [30]. Tumor secreted factors help vascular network formation, distinct from normal vasculature due to structural abnormalities, disorganized layout, increased leakiness, and aberrant osmotic forces [31][32][33][34].
The leaky tumor vasculature was shown to occur after co-culturing human ovarian cancer spheroid and endothelial cells within a dense matrix on a chip [35]. This platform predicted nanoparticle accumulation in the in vivo tumor model and provided a powerful tool for evaluating nanoparticle delivery to the tumor cells. Modeling a microvascular network on a 3D microfluidic system recapitulated in vivo histologic and biochemical features of lung and brain cancers and provided a versatile platform for testing the efficacy of anti-angiogenic drugs [36][37]. Similar approaches in hematologic malignancies were able to assess anti-angiogenic agents in individual patients [38][39]. More recently, in a breast OOC cancer model, breast cancer-driven organoids loaded into a multi-chamber microfluidic chip supported the 3D growth of angiogenic blood vessels towards the cancerous organoids [40]. By showing a reduction in tumor growth with paclitaxel’s vascular perfusion, this model also confirmed the potential use of the organoid-based device in personalized drug response ex-vivo. Together, these studies propose microfluidic chips as promising platforms for modeling angiogenesis in cancer and assessing individual responses to anti-angiogenic agents.
1.3. Metastasis
Metastasis begins when cancer cells invade the basement membrane and migrate through the tumor matrix into lymphatics or blood vessels to reach a remote site [1][41][42][43]. Different mechanical properties of each space, such as confinement and stiffness, can affect this migration [44][45][46]. The effect of confinement on cancer cell migration was successfully shown using microfluidic devices. The incorporation of cancer cells into different channels of chip devices showed that confinement alone, in the absence of any chemical gradient, can influence cancer migration [47]. Exposing cells to drugs that alter microtubule dynamics, such as Taxol, seemed to lower the migratory capability.
A large body of evidence exists to show the utility of chip devices in modeling metastasis via cell–cell interaction and cellular signaling [48]. Addition of chemokines (e.g., TNF-α) and immune cells (i.e., macrophages) to a chip device induced vascular leakiness and intravasation of tumor cells to the endothelial layer as quantified by real-time visualization [49]. Similarly, a microfluidic device was successfully employed to model bone metastasis in breast cancer, where extravasation of metastatic breast cancer cells to bone marrow-driven mesenchymal stem cells was observed [50].
Overall, these data and similar studies show the utility of microfluidic chips in modeling tumor cells within their microenvironment for better studying tumor characteristics [1][51]. Given the key involvement of the TME in tumor progression and drug response in PDAC, chips seem to be a promising platform to build individualized OOCs for PDAC. Next, we will review the challenges of modeling TME in PDAC and the promises of chip platforms.
2. OOCs to Model TME in PDAC
The TME has an active role in tumor progression, immune evasion, and drug response in PDAC [52][53]. Non-cellular components of TME of pancreas tumors are composed of ECM and dense fibrous tissue, regarded as “desmoplasia” (Figure 1) [54]. The dense ECM is mainly composed of matrix proteins such as collagen secreted by the cellular components of TME such as fibroblasts and pancreatic stellate cells (PSCs) [55]. PSCs can reduce cancer cell death upon chemotherapy induction by releasing soluble factors or activating stemness signaling pathways in cancer cells [56]. Several other immune cell types are present in the TME, among which macrophages are the most abundant that gauge both the innate and adaptive immune responses against the tumor [55]. Similar to PSCs, macrophages in interaction with cancer cells can shift towards a pro-tumorigenic phenotype, which leads to increased cancer cell stemness and growth [57]. Macrophages are also crucial in rendering chemoresistance to conventional chemotherapeutics for PDAC [58]. Pancreatic epithelial ducts secrete alkali in the epical side and create an alkaline pH in the microenvironment to prevent the breakdown of secreted pre-enzymes in the normal pancreas before reaching the small bowel lumen [59]. In PDAC, dysregulation of ion channel transporters (i.e., Ca2+ and K+ channels) and the tumor hypovascularization contribute to the acidification of the microenvironment, which further promotes cancerous characteristics in the epithelium (e.g., selection of EMT phenotypes) while shifting the stromal cells (i.e., PSC and macrophages) towards protumorigenic phenotypes [59][60]. While there are numerous other cell types in the TME (e.g., T cells, B cells, Dendritic cells), we focus on collagen-producing cells and macrophages as two major components of the TME in PDAC. In the following sections, we highlight the characteristics and plasticity of these cell types within the tumor area before discussing the opportunities provided by OOCs to model TME in PDAC.
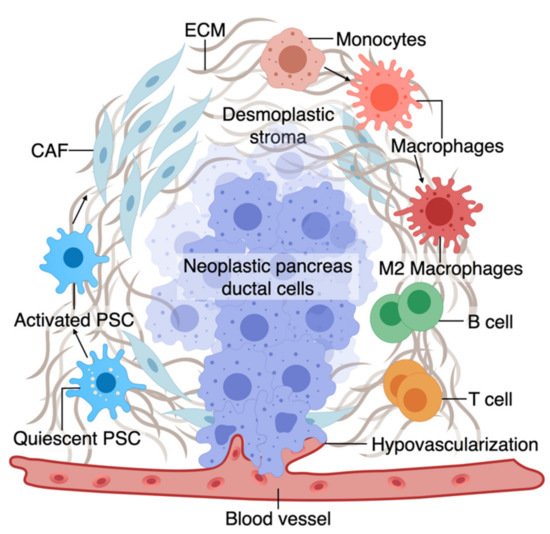
Figure 1. A. The tumor microenvironment of PDAC is composed of interactions among different cell types. At an early stage, interactions of transforming neoplastic ductal cells with pancreatic stellate cells, fibroblasts, and macrophages result in initiating a fibroinflammatory process and subsequent involvement of an adaptive immune response, including recruitment of T cells. As the tumor progresses, cancer cells invade the blood vessels and occupy the vessel lumen, referred to as endothelial ablation. Endothelial ablation and a desmoplastic stroma, accompanied by a suppressive immune environment, reduce drug delivery and chemoresistance at the site.
2.1. Macrophages and Fibroblasts in PDAC
Macrophages in the pancreatic tissue are immune cells that can arise from the embryonic precursors traced back to the extraembryonic yolk or infiltrating monocytes from myeloid precursors in the bone marrow [61][62][63][64][65][66]. The tissue macrophages can change their function (polarization) in response to surrounding signals. While macrophages were traditionally classified into M1 and M2 subtypes, mounting evidence supports the presence of a wide phenotypic spectrum between M1 with stronger killing properties to M2 that can contribute to a smoldering chronic inflammatory state in cancer [66][67][68][69][70]. M1 macrophages, through the production of nitrogen and oxygen derivatives, possess anti-tumorigenic ability by identifying and destroying cancer cells through phagocytosis [67].
In contrast, M2 macrophages can promote tumor growth via multiple mechanisms, as discussed in detail elsewhere [67][71]. While M1 macrophages could be predominant in the TME during the early stages of cancer formation, the M2-type phenotype becomes more abundant as the tumor progresses. In fact, tumor-associated macrophages are more similar to M2-types and predict poor survival [71][72][73]. Macrophage polarization or recruitment within the pancreas tissue occurs via crosstalk with cancerous epithelium and other components of the TME. In line with the growing literature on the crosstalk between macrophages and cancerous epithelium in PDAC, we also showed that early carcinogenesis signaling in the pancreatic epithelium could shift macrophages towards M2-like cells and that polarized macrophages could further promote cancer formation via induction of inflammatory signaling [74][55].
The major source of matrix deposition in the TME is cancer-associated fibroblasts (CAFs) that can arise from the PSCs, the resident mesenchymal cells in the pancreas [75][76][77]. Cancer cells activate resident PSCs, which differentiate into CAFs and secrete matrix proteins such as collagen, contributing to the dense ECM [72]. Activated PSCs also secrete factors to induce tumor growth, progression, and metastasis [73][75][78][79].
While the dense ECM has been traditionally considered to help tumor progression, recent studies support a more complex role for ECM’s contribution to PDAC progression, which could be stage and context-dependent [80][81]. However, at a late stage, the ECM contributes to tumor chemoresistance via multiple mechanisms such as cancer cell sensitivity, drug cytotoxicity, and reduced drug delivery [82][83][84]. Tumor fibroblasts are associated with poor drug response and disease survival partly by offsetting chemotherapy-induced apoptosis via soluble factors or activating stemness signaling pathways in the cancer cells [85][86][87][88][89]. Similar to macrophages, pancreatic fibroblasts could be phenotypically plastic and dynamic in response to the surrounding tumor stimuli [90][91]. Although continuous matrix deposition turns on the signaling pathways to boost the malignant phenotype, more desmoplasia contributes to tumor progression and drug resistance [92][93][94]. While the role of stroma in promoting resistance in drug response is well accepted, recent animal studies show conflicting roles of stroma in tumor formation in PDAC. Protective effects of stroma were suggested by increased tumor aggression upon stroma reduction in a mouse model where Sonic hedgehog (Shh), a soluble ligand that drives the formation of desmoplastic stroma, was deleted [95].
2.2. TME on Chip Models of PDAC
A biomimetic ductal TME on a chip, where ductal pancreatic cancer epithelium cells were surrounded by collagen matrix in the chip, recapitulated the histopathology of PDAC [96]. The tumor heterogeneity was reconstituted using pancreatic cancer cells from GEMM carrying KRAS, CDKN2A, and TP53 mutations, key driver mutations of human PDAC. This model revealed the complex interactions between cancerous epithelial cells in PDAC, leading them to be more aggressive and invasive [96].
If we are able to mimic the active TME on multicellular OOCs, can we use these platforms to model PDAC drug response in vitro? A humanized microfluidic device, where PSCs were cultured with PANC-1 cells, represented expected histologic features of PDAC and showed the potential adjuvant therapeutic activity of anti-TME agents to conventional chemotherapies [97]. The data suggest that besides studying cytotoxicity, this model has the potential to determine the effects of TME compactness and collagen reorganization on PDAC therapeutics [97]. A similar drug response to Cisplatin has been shown in a microfluidic chamber cultured with different PDAC cells in ECM-enriched environments [98].
Moving towards a patient-based ex vivo preclinical platform, work is in progress to use tissue-driven cells in OCC models of cancer. In this regard, organoids have made it possible to test drugs on the individual’s tumor cells in the lab. Next, we will discuss how the integration of organotypic technologies in OOCs could allow us to model complex cellular interactions, and the therapeutic activity of anticancer drugs, with the potential to design novel therapeutics at the individual level.
3. Individualized PDAC Model on Chip
The variation in sensitivity to anticancer drugs among different patients highlights the requirement for more precise treatment selection [99]. We and others have shown that organoids retain a high degree of similarity to the original tissue, including PDAC [74][100]. PDOs are proposed to provide an opportunity for a personalized in vitro platform to test drug sensitivity in individual patients [101]. In PDAC, organoid technology is instrumental in optimizing the use of sparse tissue collected from clinically indicated endoscopic fine needle biopsies (FNBs) performed for tissue diagnosis. This circumvents the particular challenges to precision medicine in PDAC, which stems from limited access to surgically-naïve specimens for pre-treatment screening (>80% of PDACs are unresectable) and rapid patient deterioration [102]. While organoids are superior to conventional cells for predicting drug response, they often show uncertain growth and considerable heterogeneity and are challenging to manipulate using conventional in vitro techniques [99]. Therefore, culturing them in microfluidic OOC platforms that mimic 3D tissue architecture and better facilitate nutrient and gas exchange could be more faithful in modeling the disease [103]. Work is in progress to make the organoid-based models more complex to simulate in vivo tissue structures [104][105].
Such a personalized in vitro chip model was recently developed using PDOs derived from PDAC tumor biopsy, fibroblasts, and endothelial cells tri-cultured in a perfusable 96-well based OOC system [7]. Symbiotic interaction between the PDOs and fibroblasts was observed with an elevated proliferation and increased PDO diameter in the co-culture system. Moreover, fibroblast contributed to chemoresistance to gemcitabine by secreting collagen, which added to the matrix stiffness and acted as a physical barrier to drug delivery. This co-culture platform showed the importance of the relationship between patient cells and desmoplastic ECM and provided a better understanding of chemotherapeutic agents’ bioavailability inside vascularized tumor tissues. Such a model could demonstrate a particular anticancer drug’s sensitivity to an individual patient in the lab before applying in the clinic.
This entry is adapted from the peer-reviewed paper 10.3390/cancers13174487
References
- Sontheimer-Phelps, A.; Hassell, B.A.; Ingber, D.E. Modelling cancer in microfluidic human organs-on-chips. Nat. Rev. Cancer 2019, 19, 65–81.
- Ren, K.; Zhou, J.; Wu, H. Materials for microfluidic chip fabrication. Acc. Chem. Res. 2013, 46, 2396–2406.
- Graiver, D.; Farminer, K.W.; Narayan, R. A Review of the Fate and Effects of Silicones in the Environment. J. Polym. Environ. 2003, 11, 129–136.
- Fiorini, G.S.; Chiu, D.T. Disposable microfluidic devices: Fabrication, function, and application. Biotechniques 2005, 38, 429–446.
- Prodanov, L.; Jindal, R.; Bale, S.S.; Hegde, M.; McCarty, W.J.; Golberg, I.; Bhushan, A.; Yarmush, M.L.; Usta, O.B. Long-term maintenance of a microfluidic 3D human liver sinusoid. Biotechnol. Bioeng. 2016, 113, 241–246.
- Markov, D.A.; Lillie, E.M.; Garbett, S.P.; McCawley, L.J. Variation in diffusion of gases through PDMS due to plasma surface treatment and storage conditions. Biomed Microdevices 2014, 16, 91–96.
- Lai Benjamin, F.L.; Lu Rick, X.; Hu, Y.; Davenport, H.L.; Dou, W.; Wang, E.Y.; Radulovich, N.; Tsao, M.S.; Sun, Y.; Radisic, M. Recapitulating pancreatic tumor microenvironment through synergistic use of patient organoids and organ-on-a-chip vasculature. Adv. Funct. Mater. 2020, 30, 2000545.
- Khan, O.F.; Sefton, M.V. Endothelial cell behaviour within a microfluidic mimic of the flow channels of a modular tissue engineered construct. Biomed Microdevices 2011, 13, 69–87.
- Varma, S.; Voldman, J. A cell-based sensor of fluid shear stress for microfluidics. Lab Chip 2015, 15, 1563–1573.
- Mani, V.; Lyu, Z.; Kumar, V.; Ercal, B.; Chen, H.; Malhotra, S.V.; Demirci, U. Epithelial-to-Mesenchymal Transition (EMT) and Drug Response in Dynamic Bioengineered Lung Cancer Microenvironment. Adv. Biosyst. 2019, 3, e1800223.
- Ayres Pereira, M.; Chio, I.I.C. Metastasis in Pancreatic Ductal Adenocarcinoma: Current Standing and Methodologies. Genes 2019, 11, 6.
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196.
- Diepenbruck, M.; Christofori, G. Epithelial-mesenchymal transition (EMT) and metastasis: Yes, no, maybe? Curr. Opin. Cell Biol. 2016, 43, 7–13.
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784.
- Rizvi, I.; Gurkan, U.A.; Tasoglu, S.; Alagic, N.; Celli, J.P.; Mensah, L.B.; Mai, Z.; Demirci, U.; Hasan, T. Flow induces epithelial-mesenchymal transition, cellular heterogeneity and biomarker modulation in 3D ovarian cancer nodules. Proc. Natl. Acad. Sci. USA 2013, 110, E1974–E1983.
- Calibasi Kocal, G.; Guven, S.; Foygel, K.; Goldman, A.; Chen, P.; Sengupta, S.; Paulmurugan, R.; Baskin, Y.; Demirci, U. Dynamic Microenvironment Induces Phenotypic Plasticity of Esophageal Cancer Cells Under Flow. Sci. Rep. 2016, 6, 38221.
- Armbrecht, L.; Muller, R.S.; Nikoloff, J.; Dittrich, P.S. Single-cell protein profiling in microchambers with barcoded beads. Microsyst. Nanoeng. 2019, 5, 55.
- Easley, C.J.; Karlinsey, J.M.; Bienvenue, J.M.; Legendre, L.A.; Roper, M.G.; Feldman, S.H.; Hughes, M.A.; Hewlett, E.L.; Merkel, T.J.; Ferrance, J.P.; et al. A fully integrated microfluidic genetic analysis system with sample-in-answer-out capability. Proc. Natl. Acad. Sci. USA 2006, 103, 19272–19277.
- Honegger, T.; Peyrade, D. Dielectrophoretic properties of engineered protein patterned colloidal particles. Biomicrofluidics 2012, 6, 44115.
- Roper, M.G.; Shackman, J.G.; Dahlgren, G.M.; Kennedy, R.T. Microfluidic chip for continuous monitoring of hormone secretion from live cells using an electrophoresis-based immunoassay. Anal. Chem. 2003, 75, 4711–4717.
- Luan, Q.; Cahoon, S.; Wu, A.; Bale, S.S.; Yarmush, M.; Bhushan, A. A microfluidic in-line ELISA for measuring secreted protein under perfusion. Biomed Microdevices 2017, 19, 101.
- Verma, M.S.; Tsaloglou, M.N.; Sisley, T.; Christodouleas, D.; Chen, A.; Milette, J.; Whitesides, G.M. Sliding-strip microfluidic device enables ELISA on paper. Biosens. Bioelectron. 2018, 99, 77–84.
- Zhang, Y.S.; Aleman, J.; Shin, S.R.; Kilic, T.; Kim, D.; Mousavi Shaegh, S.A.; Massa, S.; Riahi, R.; Chae, S.; Hu, N.; et al. Multisensor-integrated organs-on-chips platform for automated and continual in situ monitoring of organoid behaviors. Proc. Natl. Acad. Sci. USA 2017, 114, E2293–E2302.
- Orimo, A.; Weinberg, R.A. Stromal fibroblasts in cancer: A novel tumor-promoting cell type. Cell Cycle 2006, 5, 1597–1601.
- Ahn, J.; Sei, Y.J.; Jeon, N.L.; Kim, Y. Tumor Microenvironment on a Chip: The Progress and Future Perspective. Bioengineering 2017, 4, 64.
- Domenech, M.; Yu, H.; Warrick, J.; Badders, N.M.; Meyvantsson, I.; Alexander, C.M.; Beebe, D.J. Cellular observations enabled by microculture: Paracrine signaling and population demographics. Integr. Biol. 2009, 1, 267–274.
- Jeong, S.-Y.; Lee, J.-H.; Shin, Y.; Chung, S.; Kuh, H.-J. Co-Culture of Tumor Spheroids and Fibroblasts in a Collagen Matrix-Incorporated Microfluidic Chip Mimics Reciprocal Activation in Solid Tumor Microenvironment. PLoS ONE 2016, 11, e0159013.
- Huang, C.P.; Lu, J.; Seon, H.; Lee, A.P.; Flanagan, L.A.; Kim, H.Y.; Putnam, A.J.; Jeon, N.L. Engineering microscale cellular niches for three-dimensional multicellular co-cultures. Lab Chip 2009, 9, 1740–1748.
- Sung, K.E.; Yang, N.; Pehlke, C.; Keely, P.J.; Eliceiri, K.W.; Friedl, A.; Beebe, D.J. Transition to invasion in breast cancer: A microfluidic in vitro model enables examination of spatial and temporal effects. Integr. Biol. 2011, 3, 439–450.
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257.
- Siemann, D.W. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by Tumor-Vascular Disrupting Agents. Cancer Treat. Rev. 2011, 37, 63–74.
- Gee, M.S.; Procopio, W.N.; Makonnen, S.; Feldman, M.D.; Yeilding, N.M.; Lee, W.M. Tumor vessel development and maturation impose limits on the effectiveness of anti-vascular therapy. Am. J. Pathol. 2003, 162, 183–193.
- Tong, R.T.; Boucher, Y.; Kozin, S.V.; Winkler, F.; Hicklin, D.J.; Jain, R.K. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004, 64, 3731–3736.
- Boocock, C.A.; Charnock-Jones, D.S.; Sharkey, A.M.; McLaren, J.; Barker, P.J.; Wright, K.A.; Twentyman, P.R.; Smith, S.K. Expression of vascular endothelial growth factor and its receptors flt and KDR in ovarian carcinoma. J. Natl. Cancer Inst. 1995, 87, 506–516.
- Wang, H.-F.; Ran, R.; Liu, Y.; Hui, Y.; Zeng, B.; Chen, D.; Weitz, D.A.; Zhao, C.-X. Tumor-Vasculature-on-a-Chip for Investigating Nanoparticle Extravasation and Tumor Accumulation. ACS Nano 2018, 12, 11600–11609.
- Kim, S.; Lee, H.; Chung, M.; Jeon, N. Engineering of functional, perfusable 3D microvascular networks on a chip. Lab Chip 2013, 13.
- Lee, H.; Park, W.; Ryu, H.; Jeon, N.L. A microfluidic platform for quantitative analysis of cancer angiogenesis and intravasation. Biomicrofluidics 2014, 8, 054102.
- Ribatti, D. Is angiogenesis essential for the progression of hematological malignancies or is it an epiphenomenon? Leukemia 2009, 23, 433–434.
- Zheng, Y.; Sun, Y.; Yu, X.; Shao, Y.; Zhang, P.; Dai, G.; Fu, J. Angiogenesis in Liquid Tumors: An In Vitro Assay for Leukemic-Cell-Induced Bone Marrow Angiogenesis. Adv. Healthc. Mater. 2016, 5, 1014–1024.
- Shirure, V.S.; Bi, Y.; Curtis, M.B.; Lezia, A.; Goedegebuure, M.M.; Goedegebuure, S.P.; Aft, R.; Fields, R.C.; George, S.C. Tumor-on-a-chip platform to investigate progression and drug sensitivity in cell lines and patient-derived organoids. Lab Chip 2018, 18, 3687–3702.
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009.
- Quigley, J.P.; Armstrong, P.B. Tumor cell intravasation alu-cidated: The chick embryo opens the window. Cell 1998, 94, 281–284.
- Wolf, K.; Te Lindert, M.; Krause, M.; Alexander, S.; Te Riet, J.; Willis, A.L.; Hoffman, R.M.; Figdor, C.G.; Weiss, S.J.; Friedl, P. Physical limits of cell migration: Control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J. Cell Biol. 2013, 201, 1069–1084.
- Peyton, S.R.; Putnam, A.J. Extracellular matrix rigidity governs smooth muscle cell motility in a biphasic fashion. J. Cell Physiol. 2005, 204, 198–209.
- Hawkins, R.J.; Piel, M.; Faure-Andre, G.; Lennon-Dumenil, A.M.; Joanny, J.F.; Prost, J.; Voituriez, R. Pushing off the walls: A mechanism of cell motility in confinement. Phys. Rev. Lett. 2009, 102, 058103.
- Paul, C.D.; Hung, W.C.; Wirtz, D.; Konstantopoulos, K. Engineered Models of Confined Cell Migration. Annu. Rev. Biomed. Eng. 2016, 18, 159–180.
- Irimia, D.; Toner, M. Spontaneous migration of cancer cells under conditions of mechanical confinement. Integr. Biol. 2009, 1, 506–512.
- Nguyen, D.T.; Lee, E.; Alimperti, S.; Norgard, R.J.; Wong, A.; Lee, J.J.; Eyckmans, J.; Stanger, B.Z.; Chen, C.S. A biomimetic pancreatic cancer on-chip reveals endothelial ablation via ALK7 signaling. Sci. Adv. 2019, 5, eaav6789.
- Zervantonakis, I.K.; Hughes-Alford, S.K.; Charest, J.L.; Condeelis, J.S.; Gertler, F.B.; Kamm, R.D. Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc. Natl. Acad. Sci. USA 2012, 109, 13515–13520.
- Bersini, S.; Jeon, J.S.; Dubini, G.; Arrigoni, C.; Chung, S.; Charest, J.L.; Moretti, M.; Kamm, R.D. A microfluidic 3D in vitro model for specificity of breast cancer metastasis to bone. Biomaterials 2014, 35, 2454–2461.
- Delle Cave, D.; Rizzo, R.; Sainz, B., Jr.; Gigli, G.; Del Mercato, L.L.; Lonardo, E. The Revolutionary Roads to Study Cell-Cell Interactions in 3D In Vitro Pancreatic Cancer Models. Cancers 2021, 13, 930.
- Martinez-Bosch, N.; Vinaixa, J.; Navarro, P. Immune Evasion in Pancreatic Cancer: From Mechanisms to Therapy. Cancers 2018, 10, 6.
- Beatty, G.L.; Gladney, W.L. Immune Escape Mechanisms as a Guide for Cancer Immunotherapy. Clin. Cancer Res. 2015, 21, 687–692.
- Pandol, S.; Edderkaoui, M.; Gukovsky, I.; Lugea, A.; Gukovskaya, A. Desmoplasia of pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 2009, 7 (Suppl. 11), S44–S47.
- Pandol, S.J.; Edderkaoui, M. What are the macrophages and stellate cells doing in pancreatic adenocarcinoma? Front. Physiol. 2015, 6, 125.
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276.
- Cui, R.; Yue, W.; Lattime, E.C.; Stein, M.N.; Xu, Q.; Tan, X.L. Targeting tumor-associated macrophages to combat pancreatic cancer. Oncotarget 2016, 7, 50735–50754.
- Amit, M.; Gil, Z. Macrophages increase the resistance of pancreatic adenocarcinoma cells to gemcitabine by upregulating cytidine deaminase. Oncoimmunology 2013, 2, e27231.
- Pedersen, S.F.; Novak, I.; Alves, F.; Schwab, A.; Pardo, L.A. Alternating pH landscapes shape epithelial cancer initiation and progression: Focus on pancreatic cancer. Bioessays 2017, 39.
- Ling, Q.; Kalthoff, H. Transportome Malfunctions and the Hallmarks of Pancreatic Cancer. Rev. Physiol. Biochem. Pharmacol. 2020.
- Hoeffel, G.; Ginhoux, F. Ontogeny of Tissue-Resident Macrophages. Front. Immunol. 2015, 6, 486.
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845.
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128.
- Hoeffel, G.; Wang, Y.; Greter, M.; See, P.; Teo, P.; Malleret, B.; Leboeuf, M.; Low, D.; Oller, G.; Almeida, F.; et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J. Exp. Med. 2012, 209, 1167–1181.
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90.
- Guilliams, M.; De Kleer, I.; Henri, S.; Post, S.; Vanhoutte, L.; De Prijck, S.; Deswarte, K.; Malissen, B.; Hammad, H.; Lambrecht, B.N. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med. 2013, 210, 1977–1992.
- Rhee, I. Diverse macrophages polarization in tumor microenvironment. Arch. Pharm. Res. 2016, 39, 1588–1596.
- Elliott, M.R.; Koster, K.M.; Murphy, P.S. Efferocytosis Signaling in the Regulation of Macrophage Inflammatory Responses. J. Immunol. 2017, 198, 1387–1394.
- Taylor, P.R.; Martinez-Pomares, L.; Stacey, M.; Lin, H.H.; Brown, G.D.; Gordon, S. Macrophage receptors and immune recognition. Annu. Rev. Immunol. 2005, 23, 901–944.
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964.
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126.
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596.
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926.
- Bishehsari, F.; Zhang, L.; Barlass, U.; Preite, N.Z.; Turturro, S.; Najor, M.S.; Shetuni, B.B.; Zayas, J.P.; Mahdavinia, M.; Abukhdeir, A.M.; et al. KRAS mutation and epithelial-macrophage interplay in pancreatic neoplastic transformation. Int. J. Cancer 2018, 143, 1994–2007.
- Barros, M.H.; Segges, P.; Vera-Lozada, G.; Hassan, R.; Niedobitek, G. Macrophage polarization reflects T cell composition of tumor microenvironment in pediatric classical Hodgkin lymphoma and has impact on survival. PLoS ONE 2015, 10, e0124531.
- Erkan, M.; Adler, G.; Apte, M.V.; Bachem, M.G.; Buchholz, M.; Detlefsen, S.; Esposito, I.; Friess, H.; Gress, T.M.; Habisch, H.J.; et al. StellaTUM: Current consensus and discussion on pancreatic stellate cell research. Gut 2012, 61, 172–178.
- Apte, M.V.; Wilson, J.S.; Lugea, A.; Pandol, S.J. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology 2013, 144, 1210–1219.
- Vonlaufen, A.; Phillips, P.A.; Xu, Z.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Pancreatic stellate cells and pancreatic cancer cells: An unholy alliance. Cancer Res. 2008, 68, 7707–7710.
- Xu, Z.; Vonlaufen, A.; Phillips, P.A.; Fiala-Beer, E.; Zhang, X.; Yang, L.; Biankin, A.V.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am. J. Pathol. 2010, 177, 2585–2596.
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505.
- Gore, J.; Korc, M. Pancreatic cancer stroma: Friend or foe? Cancer Cell 2014, 25, 711–712.
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234.
- Schober, M.; Jesenofsky, R.; Faissner, R.; Weidenauer, C.; Hagmann, W.; Michl, P.; Heuchel, R.L.; Haas, S.L.; Lohr, J.M. Desmoplasia and chemoresistance in pancreatic cancer. Cancers 2014, 6, 2137–2154.
- Ireland, L.; Santos, A.; Ahmed, M.S.; Rainer, C.; Nielsen, S.R.; Quaranta, V.; Weyer-Czernilofsky, U.; Engle, D.D.; Perez-Mancera, P.A.; Coupland, S.E.; et al. Chemoresistance in Pancreatic Cancer Is Driven by Stroma-Derived Insulin-Like Growth Factors. Cancer Res. 2016, 76, 6851–6863.
- Zhang, H.; Wu, H.; Guan, J.; Wang, L.; Ren, X.; Shi, X.; Liang, Z.; Liu, T. Paracrine SDF-1alpha signaling mediates the effects of PSCs on GEM chemoresistance through an IL-6 autocrine loop in pancreatic cancer cells. Oncotarget 2015, 6, 3085–3097.
- Erkan, M.; Reiser-Erkan, C.; Michalski, C.W.; Deucker, S.; Sauliunaite, D.; Streit, S.; Esposito, I.; Friess, H.; Kleeff, J. Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia 2009, 11, 497–508.
- Ide, T.; Kitajima, Y.; Miyoshi, A.; Ohtsuka, T.; Mitsuno, M.; Ohtaka, K.; Miyazaki, K. The hypoxic environment in tumor-stromal cells accelerates pancreatic cancer progression via the activation of paracrine hepatocyte growth factor/c-Met signaling. Ann. Surg. Oncol. 2007, 14, 2600–2607.
- Duluc, C.; Moatassim-Billah, S.; Chalabi-Dchar, M.; Perraud, A.; Samain, R.; Breibach, F.; Gayral, M.; Cordelier, P.; Delisle, M.B.; Bousquet-Dubouch, M.P.; et al. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol. Med. 2015, 7, 735–753.
- Watanabe, I.; Hasebe, T.; Sasaki, S.; Konishi, M.; Inoue, K.; Nakagohri, T.; Oda, T.; Mukai, K.; Kinoshita, T. Advanced Pancreatic Ductal Cancer: Fibrotic Focus and β-Catenin Expression Correlate With Outcome. Pancreas 2003, 26, 326–333.
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598.
- Pure, E.; Hingorani, S.R. Mesenchymal Cell Plasticity and Perfidy in Epithelial Malignancy. Trends Cancer 2018, 4, 273–277.
- Merika, E.E.; Syrigos, K.N.; Saif, M.W. Desmoplasia in pancreatic cancer. Can we fight it? Gastroenterol. Res. Pract. 2012, 2012, 781765.
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400.
- Ghajar, C.M.; Chen, X.; Harris, J.W.; Suresh, V.; Hughes, C.C.; Jeon, N.L.; Putnam, A.J.; George, S.C. The effect of matrix density on the regulation of 3-D capillary morphogenesis. Biophys. J. 2008, 94, 1930–1941.
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747.
- Bradney, M.J.; Venis, S.M.; Yang, Y.; Konieczny, S.F.; Han, B. A Biomimetic Tumor Model of Heterogeneous Invasion in Pancreatic Ductal Adenocarcinoma. Small 2020, 16, e1905500.
- Rhim, A.D.; Thege, F.I.; Santana, S.M.; Lannin, T.B.; Saha, T.N.; Tsai, S.; Maggs, L.R.; Kochman, M.L.; Ginsberg, G.G.; Lieb, J.G.; et al. Detection of circulating pancreas epithelial cells in patients with pancreatic cystic lesions. Gastroenterology 2014, 146, 647–651.
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361.
- Drifka, C.R.; Eliceiri, K.W.; Weber, S.M.; Kao, W.J. A bioengineered heterotypic stroma-cancer microenvironment model to study pancreatic ductal adenocarcinoma. Lab Chip 2013, 13, 3965–3975.
- Beer, M.; Kuppalu, N.; Stefanini, M.; Becker, H.; Schulz, I.; Manoli, S.; Schuette, J.; Schmees, C.; Casazza, A.; Stelzle, M.; et al. A novel microfluidic 3D platform for culturing pancreatic ductal adenocarcinoma cells: Comparison with in vitro cultures and in vivo xenografts. Sci. Rep. 2017, 7, 1325.
- Weeber, F.; van de Wetering, M.; Hoogstraat, M.; Dijkstra, K.K.; Krijgsman, O.; Kuilman, T.; Gadellaa-van Hooijdonk, C.G.; van der Velden, D.L.; Peeper, D.S.; Cuppen, E.P.; et al. Preserved genetic diversity in organoids cultured from biopsies of human colorectal cancer metastases. Proc. Natl. Acad. Sci. USA 2015, 112, 13308–13311.
- Armstrong, A.H.M.R.; Mirbagheri, S.; Barlass, U.; Gilbert, D.Z.; Amin, J.; Singh, A.; Naqib, A.; Bishehsari, F. Multiplex Patient-Based Drug Response Assay in Pancreatic Ductal Adenocarcinoma. Biomedicines 2021, 9, 705.
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945.
- Zins, M.; Matos, C.; Cassinotto, C. Pancreatic Adenocarcinoma Staging in the Era of Preoperative Chemotherapy and Radiation Therapy. Radiology 2018, 287, 374–390.
- Yu, F.; Hunziker, W.; Choudhury, D. Engineering Microfluidic Organoid-on-a-Chip Platforms. Micromachines 2019, 10, 165.
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418.
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379.