Depsides are a group of polyketides consisting of two or more ester-linked hydroxybenzoic acid moieties. They possess valuable bioactive properties, such as anticancer, antidiabetic, antibacterial, antiviral, anti-inflammatory, antifungal, antifouling, and antioxidant qualities, as well as various human enzyme-inhibitory activities.
- fungi
- depsides
- biosynthesis
- spectral data
- biological activities
1. Introduction
Depsides are simple polyketides that are formed by the condensation of two or more hydroxybenzoic acid moieties via ester linkage; the COOH group of one molecule is esterified with a phenolic OH group of the second molecule. They could be β-orcinol (β-orsellinic acid) or orcinol (orsellinic acid) derivatives, relying on the existence of the C3 methyl group on both rings (Figure 1). The ring with an ester-carbonyl is referred to as ring A and the other as ring B. Their major structural variations are the attached alkyl chains’ length, the degree of chain oxidation, and the degree of methylation of OH and COOH groups [18]. The OH groups usually exist at the aromatic carbons, C-3′/C-4′/C-2 or C-4, and other oxygenated substituents are usually connected to the skeleton, such as carboxyl and methoxyl substituents [19].
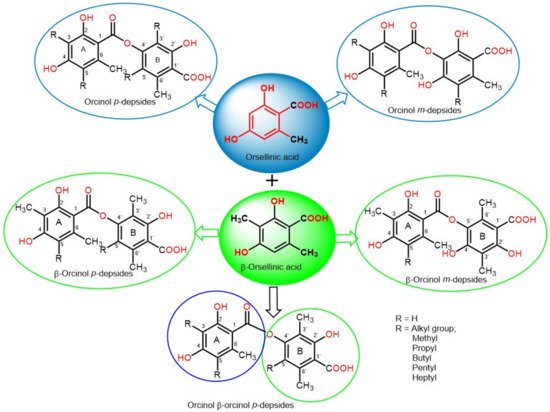
2. Biosynthesis of Depsides
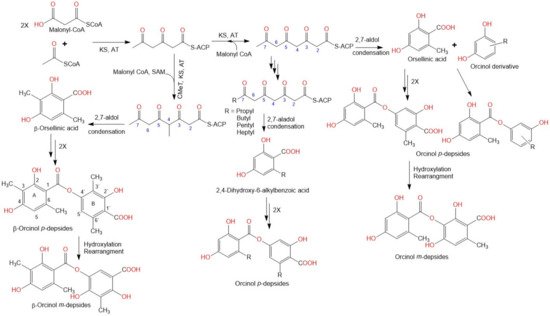
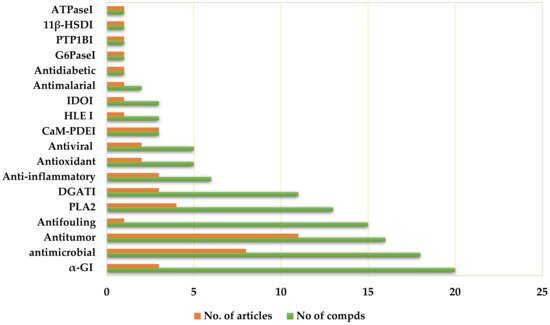
3. Biological Activities
3.1. Antitumor Activity
Lünne et al. [37] evaluated the antitumor effect of lecanoric acid (1) and ethyl lecanorate (2) purified from Claviceps purpurea on HepG2 (human liver cancer cells) and CCF-STTG1 (human astrocytoma cells) using the CTC (5-cyano-2,3-bis(4-methylphenyl)-2H-tetrazolium chloride) assay. Both metabolites produced a dose-dependent antitumor effect on the tested cell lines. They reduced the CCF-STTG1 cell viability down to ~60%, at a concentration of 40 μM, and HepG2 cell viability by ~30% and 40%, respectively. Similar to HepG2 cells, 2 had the strongest antitumor effect on CCF cells (IC50 value of 54 μM) [37]. In the MTT ((3-(4,5-dimethylthiazol-2-yl))-2,5-diphenyl-2H-tetrazolium bromide) assay, aspergisides A (3), B (4), and C (5) showed weak antitumor activity, with IC50 values in the range of 45–114 µM toward Vero, MCF-7, and KB cell lines, compared with doxorubicin [38]. MS-3 (22) was inactive against Ehrlich ascites for leukemia and carcinoma in vivo; however, it was active toward Yoshida sarcoma cells (ID5o value of 85 µg/mL) in vitro. Its activity was suggested to be due to a glyoxalase inhibition, as it possessed a glyoxalase inhibitory effect with an ID50 value of 12 µg/mL in the spectrophotometric assay [39]. In addition, 23 possessed significant antitumor activity toward A549 and HepG2, with IC50 values of 13.14 and 49.02 µM, respectively, compared to cisplatin (IC50 14.33 and 18.74, respectively) in the MTT assay [40]. Compounds 27–29 were assayed against NCI-H187, Vero, BC, and KB cells, employing an MTT assay. Compounds 27 and 28 exhibited a significant antitumor effect against BC, with IC50 values of 8.8 and 4.4 µM, respectively, compared to ellipticine (IC50 0.49 µM), while they showed weak to moderate effectiveness toward other cell lines, with IC50 values ranging from 13.0 to 34.3 µM [41]. Arenicolins A (30) and B (31), two new depsides having C-glycosyl moiety and dual heptyl side-chains, were isolated from Penicillium arenicola and assessed for antitumor activity at a concentration of 30.0 μM toward IMR-32, HCT-116, and BT-474 cell lines using an ICC (immunocytochemistry) assay. Compound 30 reduced cell viability with IC50 values of 6.0, 7.3, and 9.7 μM, respectively, compared to 5-FU (5-fluorouracil, IC50 6.5 μM for HCT-116 and 5.7 μM for IMR-32). However, 31 did not have a significant antitumor effect toward the tested cell lines at a concentration of > 30 μM [42].
3.2. Antimicrobial Activity
Phainuphong et al. purified three new depsides, aspergisides A–C (3–5) from Aspergillus unguis, and assessed their antimicrobial potential against MRSA (methicillin-resistant S. aureus), S. aureus, C. albicans, flucytosine-resistant C. neoformans, and M. gypseum [38]. Compound 3 had a weak antibacterial activity toward S. aureus and MRSA, with an MIC (minimal inhibitory concentrations) value of 8 µg/mL, while 4 and 5 were inactive, with MIC values of 32–200 µg/mL in the agar diffusion method [38]. Setophoma sp. associated with guava fruits produced compounds 6–8 and 66–69 [19]. They did not have growth inhibition activity toward E. coli. However, 66–69 demonstrated inhibition of S. aureus with MIC values of 100, 6.25, 50, and 25 µg/mL, respectively, in comparison to tetracycline (MIC 3.12 µg/mL) [19]. Compounds 6 and 7 were inactive. Moreover, all compounds did not exhibit quorum-sensing inhibitory activity. Studying the structural activity relationship revealed that the activity increased with the full methylation of the B-ring; however, the additional CH3 group at ring A, especially at C-2, resulted in a decrease in activity [19]. The agonodepsides A (12) and B (13) were isolated from the filamentous fungus, F7524 [50]. In the fluorometric InhA assay, 12 inhibited M. tuberculosis InhA with an IC50 value of 75 µM, while 13 was inactive at 100 µM, compared with triclosan (IC50 3.0 µM) [50].
3.3. Antifouling Activity
3.4. Anti-Diabetic Activity
3.5. D-Glucose-6-Phosphate Phosphohydrolase Inhibitory Activity
3.6. α-Glucosidase Inhibitory (αGI) Activity
The α-glucosidase enzyme is an important therapeutic target for treating carbohydrate-mediated diseases. It catalyzes the breakdown of oligo- and disaccharides into monosaccharides in the final stage of carbohydrate digestion, leading to a rise in glucose levels [94,95,96,97]. Several studies revealed that α-glucosidase inhibitors (αGIs) slow down the digestion and absorption of carbohydrates, and thus reduce the postprandial blood glucose level [94,95,96,97]. The serious side effects of the current αGIs, such as liver injuries and gastrointestinal damage, have directed research efforts toward discovering and developing new and safer anti-diabetic agents.
3.7. Protein Tyrosine Phosphatase Inhibitory (PTP1BI) Activity
3.8. Diacylglycerol Acyltransferase Inhibitory (DGATI) Activity
Postprandial hypertriglyceridemia is considered the main risk factor for cardiovascular functions. Thus, triglyceride synthesis inhibition has remarkable therapeutic potential in metabolic disorder treatment. The enzymes known as diacylglycerol acyltransferases (DGATs) catalyze the final and only committed step in the biosynthesis of triglycerides [99]. Therefore, these enzymes could be a potential therapeutic target to combat cardio-metabolic disorders [71,82,99]. Compound 9 also inhibited TG synthesis (IC50 91 µM), as well as PC and PE syntheses, indicating that it had a non-specific DGATI effect [76]. The compounds 86 and 88–90 were purified from Humicola sp. by Tomoda et al. [74]. Compound 88 was the most potent DGATI, with an IC50 of 10.2 µM, followed by 86 (IC50 17.5 µM), 89 (IC50 19.2 µM), and 90 (IC50 51.6 µM). They also inhibited the formation of triacylglycerol using Raji cells on the intact cell assay, with IC50 values ranging from 2.82 to 17.2 µM. At high concentrations, 86 moderately inhibited the formation of phosphatidylethanolamine (PE) and phosphatidylcholine (PC), whereas 89 possessed a weak effect, indicating that 89 specifically suppressed the formation of triacylglycerol (TG) [74].
3.9. Activity of 11β-Hydroxysteroid Dehydrogenase Inhibitory (11β-HSDI) Enzyme
3.10. Anti-Inflammatory Activities
Compound 23, biosynthesized by Stereum hirsutum, exhibited noticeable NO inhibitory potential (IC50 19.17 µM) in the LPS-induced macrophages, compared with hydrocortisone (IC50 48.15 µM) [40]. Moreover, 48 (ID50 12 µM) and 49 (ID50 9 µM) possessed considerable anti-inflammatory potential for the conversion of 14C-arachidonic acid into PGF2α plus PGE2 by the microsomes of ram seminal vesicles [69,81]. ID50s of the conversion of arachidonic acid (AA) into PGH2 (prostaglandin H2), PGH2 into (prostaglandin E2), and thromboxane A2 (TXA2) synthetase are 10, 40, 150 µM, respectively, for 48 in comparison to indomethacin (ID50 30 for PGH2 and 130 µM for PGE2) and imidazole (ID50 200 µM for TXA2 synthetase); meanwhile, 49 had ID50 values of 40, 9, and 350 µM, respectively. Compound 48 had a strong inhibitory effect on the conversion of AA into PGH2, while 49 specifically inhibited the step involving PGE2 synthesis from PGH2. Moreover, they inhibited TXA2 synthesis in bovine platelet microsomes (ID50 values of 150 and 350 µM, respectively), which was comparable to imidazole (200 µM) [69,81]. Both compounds (dose 50 mg/kg, orally) showed no significant anti-inflammatory effects on carrageenan-induced edema in rats. However, 49 caused a 70% inhibition of this edema system at an intravenous (IV) dose of 5 mg/kg, while 48 displayed no activity, even with IV administration [81].
3.11. Antimalarial Activity
3.12. Antioxidant Activity
3.13. Ca2+/CaM Dependent Phosphodiesterase Inhibitory (CaM-PDEI) Activity
Nakanishi et al. reported that 34 and 35, purified from Sporothrix sp., inhibited heart and bovine brain PDEs (IC50 4.3 and 1.8 µM and 5.9 and 15.0 µM, respectively) [63]. Moreover, they prohibited the CaM-dependent activities of CaM-PDEs but had a low effect against their CaM-independent effects, suggesting that these compounds interacted with CaM to inhibit Ca2+/CaM-dependent enzymes. On the other hand, they had no inhibitory activities on protein kinase C [63]. Moreover, PS-990 (47), isolated from Acremonium sp., inhibited brain CaM-PDE with an IC50 value of 3 µg/mL and did not elevate the intracellular cyclic AMP level. It markedly induced the neurite extension of Neuro2A (mouse neuroblastoma) at concentrations ranging from 10 to 30 µg/mL, suggesting its neuritogenic effect. It inhibited both cell growth and thymidine incorporation into the cells at the same concentration range. Interestingly, 47 reversibly induced neurite formation, with cell growth arrest through a mechanism other than increasing the intracellular cyclic AMP concentration [66,110].
3.14. Antiviral Activity
3.15. Human Leukocyte Elastase (HLE) Inhibitory Activity
3.16. Indoleamine 2,3-Dioxygenase Inhibitory (IDOI) Activity
3.17. Adenosine Triphosphatase Inhibitory Activity
3.18. Proteasome Inhibitory Activity
3.19. Phospholipase Inhibitory Activities
Phospholipase A2 (PLA2) catalyzes the hydrolysis of membrane phospholipids into arachidonic acid; therefore, its inhibitors have the potential for treating various inflammatory disorders [117]. Compound 107 showed a strong reversible and noncompetitive inhibition of human PLA2-II (Ki value of 0.098 μM, IC50 value of 0.076 μM); however, it showed weak inhibition of human PLA2-I (IC50 of 18 μM). Its inhibitory effect toward PLA2-II human and PLA2 Naja mocambique was noticeably reduced by methylation of the two COOH groups. Furthermore, 107, upon co-injection with carrageenan, remarkably reduced PLA2 activity and exudate volume in the carrageenan-induced pleurisy rat model [80].
4. Conclusions
This entry is adapted from the peer-reviewed paper 10.3390/metabo11100683