Antisense peptide technology (APT) is based on a useful heuristic algorithm for rational peptide design. It was deduced from empirical observations that peptides consisting of complementary (sense and antisense) amino acids interact with higher probability and affinity than the randomly selected ones. This phenomenon is closely related to the structure of the standard genetic code table, and at the same time, is unrelated to the direction of its codon sequence translation.
- antisense
- complementary
- peptide
- binding
- genetic code
- bioengineering
1. Introduction
The concept of sense and antisense (i.e., complementary) peptide interaction was developed in the early 1980s by Root-Bernstein, Biro, Blalock, Mekler, Siemion, and others [1][2][3][4][5][6][7][8][9][10][11][12][13][14][15][16][17][18][19][20]. First, it was theoretically assumed and later empirically observed that peptides consisting of amino acids specified by sense and antisense sequences interact with higher probability and affinity than randomly selected peptides ( Table 1 and Table 2 , Figure 1 ). This approach was successfully applied to the investigations of more than 50 ligand–acceptor (receptor) systems, including the immune response to viral subunits and related manipulations with an epitope and paratope design [1][2][3][4][5][6][7][8][9][10][11][12][13][14][15][16][17][18][19][20][21][22].
Table 1. Standard genetic code table.
| First Letter (5′) | Second Letter | Third Letter (3′) | |||
|---|---|---|---|---|---|
| U | C | A | G | ||
| U | F | S | Y | C | U |
| F | S | Y | C | C | |
| L | S | stop | stop | A | |
| L | S | stop | W | G | |
| C | L | P | H | R | U |
| L | P | H | R | C | |
| L | P | Q | R | A | |
| L | P | Q | R | G | |
| A | I | T | N | S | U |
| I | T | N | S | C | |
| I | T | K | R | A | |
| M | T | K | R | G | |
| G | V | A | D | G | U |
| V | A | D | G | C | |
| V | A | E | G | A | |
| V | A | E | G | G | |
Table 2. Direction of translation specifies amino acid pairing.
| Amino Acid | Antisense 3′ → 5′ | Antisense 5′ → 3′ | Consensus |
|---|---|---|---|
| F | K | K, E | K |
| L | D, E, N | E, Q, K | E |
| I | Y | N, D, Y | Y |
| M | Y | H | |
| V | H, Q | H, D, N, Y | H |
| S | S, R | G, R, T, A | R |
| P | G | G, W, R | G |
| T | W, C | G, S, C, R | C |
| A | R | R, G, S, C | R |
| Y | M, I | I, V | I |
| H | V | V, M | V |
| Q | V | L | |
| N | L | I, V | |
| K | F | F, L | F |
| D | L | I, V | L |
| E | L | L, F | L |
| C | T | T, A | T |
| W | T | P | |
| R | A, S | A, S, P, T | A, S |
| G | P | P, S, T, A | P |
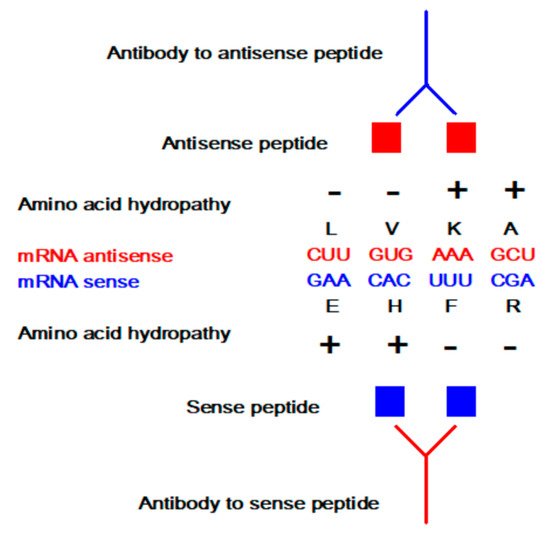
Sense peptides are essential and specific parts of viral and other proteins that elicit normal and pathologic immune responses [6][7][8][9][14][19][20][21]. Using antisense peptide technology, they could be utilized to derive targeted tests for different antibody (Ab), hormone, growth factor, or cell subpopulations [4][5][6][7][8][9][13][14][17][18][19][20][21][22][23]. The potential of antisense peptides is twofold: 1. as future diagnostic tests targeting protein epitopes or paratopes of interest, or 2. as future therapeutic agents that target specific parts of antigens to selectively modify host immune response (e.g., an antisense peptide may disrupt or modify different factors like virulence, replication or host defense) [6][7][8][9][13][14][18][19][20][21]. Consequently, sense-antisense peptide interactions may serve as a useful starting point for: 1. the development of biochemical assays for the evaluation of the immune response, and 2. modeling and design of new peptide binders for specific proteins and their receptors.
2. Antisense Peptide Technology (APT)
-
reliance on continuous epitopes,
-
overconfidence in ligand specificity,
-
amino acid bias in characterizing ligand-acceptor (receptor) interactions,
-
difficulties in the estimation of structure-function relationships between specific ligand–acceptor (receptor) pairs,
-
amino acid coding, complementarity, and frameshifts.
2.1. Reliance on Continuous Epitopes
2.2. Overconfidence in the Ligand Specificity
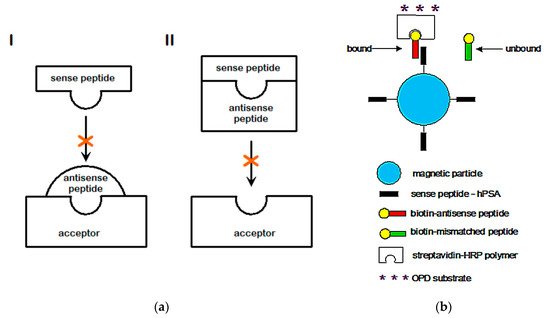
2.3. Amino Acid Bias in Characterizing Ligand-Acceptor (Receptor) Interactions
2.4. Difficulties in the Estimation of Structure-Function Relationships between Specific Ligand-Acceptor (Receptor) Pairs
-
peptides binding into molecular complexes (leaving none or low levels of sense peptide to elicit its own biological effects),
-
total or partial antagonization of the sense peptide receptor by means of its complexation with an antisense ligand,
-
combination of the first two factors,
-
other biological or biochemical effects of an antisense peptide that cannot be explained by the involvement of a sense peptide and its receptors (e.g., generation of bioactive antibodies to peptides and/or their complexes, cellular receptor, and growth factor modulation).
2.5. Amino Acid Coding, Complementarity, and Frameshifts
| Parameter | Polarity (20 aa) |
Polarity (X0, 12 aa) |
Diversity (X1, 11 aa) |
|---|---|---|---|
| GUA—nucleobase preference | −0.54 * | −0.63 * | 0.71 * |
| PUR—nucleobase preference | −0.07 | −0.49 * | 0.82 * |
| PYR—nucleobase preference | 0.06 | 0.49 * | –0.85 * |
3. Perspective
-
selection of different targets and evaluation of complementary (sense–antisense) peptide binding;
-
quantification of specific antibodies, peptides, and proteins;
-
design of MPEIAs and Multiplex ELISAs tailored for a specific purpose.
-
Quick design and validation of the complementary ligands and acceptors;
-
Computational validation and virtual screening of different protein and peptide structures;
-
Rationalization of peptide library screening;
-
The tests can be produced in a short period of time;
-
The tests will be made composite (according to the LEGO principle) and will consist of less expensive and commercially available components;
-
The time required to obtain results is shorter (since no antibody production is needed);
-
The test enables large quantity sample testing using standard laboratory equipment (since it does not require special reagents or complicated sampling processing);
-
The tests are likely to prove important for the investigation of the immune response, disease pathogenesis, and clinical outcome of different infections;
-
Designed antisense peptides (and anti-antisenses [21]) may also provide a basis for further development of vaccines and lead compounds for different diseases;
-
Detection of mutant strains is quicker since new antisense peptide motifs could be synthesized, evaluated for binding, and easily linked to magnetic particles in a short period of time, which avoids the antibody production process;
-
A green chemistry approach significantly reduces or avoids the loss of animal life.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22179106
References
- Root-Bernstein, R.S. Amino acid pairing. J. Theor. Biol. 1982, 94, 885–894.
- Loo, J.A.; Holsworth, D.D.; Root-Bernstein, R.S. Use of electrospray ionization mass spectrometry to probe antisense peptide interactions. Biol. Mass Spectrom. 1994, 23, 6–12.
- Holsworth, D.D.; Kiely, J.S.; Root-Bernstein, R.S.; Overhiser, R.W. Antisense-designed peptides: A comparative study focusing on possible complements to angiotensin II. Pept. Res. 1994, 7, 185–193.
- Root-Bernstein, R.S.; Holsworth, D.D. Antisense peptides: A critical mini-review. J. Theor. Biol. 1998, 21, 107–119.
- Root-Bernstein, R.S. Peptide self-aggregation and peptide complementarity as bases for the evolution of peptide receptors: A review. J. Mol. Recognit. 2005, 18, 40–49.
- Root-Bernstein, R. How to make a non-antigenic protein (auto) antigenic: Molecular complementarity alters antigen processing and activates adaptive-innate immunity synergy. Anticancer Agents Med. Chem. 2015, 15, 1242–1259.
- Blalock, J.E. Complementarity of peptides specified by ‘sense’ and ‘antisense’ strands of DNA. Trends. Biotechnol. 1990, 8, 140–144.
- Blalock, J.E. Genetic origin of protein shape and interaction rules. Nat. Med. 1995, 1, 876–878.
- Biro, J.C. The proteomic code: A molecular recognition code for proteins. Theor. Biol. Med. Model. 2007, 4, 1–45.
- Mekler, L.B. Specific selective interaction between amino acid residues of the polypeptide chains. Biophys. USSR 1970, 14, 613–617.
- Mekler, L.B.; Idlis, R.G. Construction of models of three-dimensional biological polypeptide and nucleoprotein molecules in agreement with a general code which determines specific linear recognition and binding of amino acid residues of polypeptides to each other and to the trinucleotides of polynucleotides. Depos. Doc. VINITI 1981, 1476–1481. (In Russian)
- Tropsha, A.; Kizert, J.S.; Chaiken, I.M. Making sense from antisense: A review of experimental data and developing ideas on sense-antisense peptide recognition. J. Mol. Recognit. 1992, 5, 43–54.
- Siemion, I.Z.; Cebrat, M.; Kluczyk, A. The problem of amino acid complementarity and antisense peptides. Curr. Protein Pept. Sci. 2004, 5, 507–527.
- Heal, J.R.; Roberts, G.W.; Raynes, J.G.; Bhakoo, A.; Miller, A.D. Specific interactions between sense and complementary peptides: The basis for the proteomic code. ChemBioChem 2002, 3, 136–151.
- Miller, A.D. Sense-antisense (complementary) peptide interactions and the proteomic code; potential opportunities in biology and pharmaceutical science. Expert. Opin. Biol. Ther. 2015, 15, 245–267.
- Štambuk, N. On the genetic origin of complementary protein coding. Croat. Chem. Acta 1998, 71, 573–589.
- Štambuk, N.; Konjevoda, P.; Boban-Blagaić, A.; Pokrić, B. Molecular recognition theory of the complementary (antisense) peptide interactions. Theory Biosci. 2005, 123, 265–275.
- Štambuk, N.; Manojlović, Z.; Turčić, P.; Martinić, R.; Konjevoda, P.; Weitner, T.; Wardega, P.; Gabričević, M. A simple three-step method for design and affinity testing of new antisense peptides: An Example of Erythropoietin. Int. J. Mol. Sci. 2014, 15, 9209–9223.
- Štambuk, N.; Konjevoda, P.; Turčić, P.; Kövér, K.; Novak Kujundžić, R.; Manojlović, Z.; Gabričević, M. Genetic coding algorithm for sense and antisense peptide interactions. Biosystems 2018, 164, 199–216.
- Štambuk, N.; Konjevoda, P.; Turčić, P.; Šošić, H.; Aralica, G.; Babić, D.; Seiwerth, S.; Kaštelan, Ž.; Kujundžić, R.N.; Wardega, P.; et al. Targeting Tumor Markers with Antisense Peptides: An Example of Human Prostate Specific Antigen. Int. J. Mol. Sci. 2019, 20, 2090.
- McGuire, K.L.; Holmes, D.S. Role of complementary proteins in autoimmunity: An old idea re-emerges with new twists. Trends Immunol. 2005, 26, 367–372.
- Dayhoff, G.W.; van Regenmortel, M.H.V.; Uversky, V.N. Intrinsic disorder in protein sense-antisense recognition. J. Mol. Recognit. 2020, 33, e2868.
- Štambuk, N.; Kopjar, N.; Šentija, K.; Garaj-Vrhovac, V.; Vikić-Topić, D.; Marušić-Della Marina, B.; Brinar, V.; Trbojević-Čepe, M.; Žarković, N.; Ćurković, B.; et al. Cytogenetic effects of met-enkephalin (peptid-M) on human lymphocytes. Croat. Chem. Acta 1998, 71, 591–605.
- Štambuk, N.; Konjevoda, P. Determining amino acid scores of the genetic code table: Complementarity, structure, function and evolution. Biosystems 2020, 187, 104026.
- Van Regenmortel, M.H.V. Synthetic peptide vaccines and the search for neutralization B cell epitopes. Open Vaccine J. 2009, 2, 33–44.
- Uversky, V.N.; van Regenmortel, M.H.V. Mobility and disorder in antibody and antigen binding sites do not prevent immunochemical recognition. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 149–156.
- Edmundson, A.B.; Ely, K.R.; Herron, J.N.; Cheson, B.D. The binding of opioid peptides to the Mcg light chain dimer: Flexible keys and adjustable locks. Mol. Immunol. 1987, 24, 915–935.
- Ciemny, M.; Kurcinski, M.; Kamel, K.; Kolinski, A.; Alam, N.; Schueler-Furman, O.; Kmiecik, S. Protein-peptide docking: Opportunities and challenges. Drug Discov. Today 2018, 23, 1530–1537.
- Pomplun, S.; Jbara, M.; Quartararo, A.J.; Zhang, G.; Brown, J.S.; Lee, Y.C.; Ye, X.; Hanna, S.; Pentelute, B.L. De novo discovery of high-affinity peptide binders for the SARS-CoV-2 spike protein. ACS Cent. Sci. 2021, 7, 156–163.
- Pomplun, S. Targeting the SARS-CoV-2-spike protein: From antibodies to miniproteins and peptides. RSC Med. Chem. 2021, 12, 197–202.
- Bowen, J.; Schneible, J.; Bacon, K.; Labar, C.; Menegatti, S.; Rao, B.M. Screening of yeast display libraries of enzymatically treated peptides to discover macrocyclic peptide ligands. Int. J. Mol. Sci. 2021, 22, 1634.
- Turčić, P.; Štambuk, N.; Konjevoda, P.; Kelava, T.; Gabričević, M.; Stojković, R.; Aralica, G. Modulation of γ2-MSH hepatoprotection by antisense peptides and melanocortin subtype 3 and 4 receptor antagonists. Med. Chem. 2015, 11, 286–925.
- Graham, P. Instant Notes in Medicinal Chemistry, 1st ed.; Taylor & Francis: London, UK, 2001; pp. 62–69.
- Woese, C.R.; Dugre, D.H.; Saxinger, W.C.; Dugre, S.A. The molecular basis for the genetic code. Proc. Natl. Acad. Sci. USA 1966, 55, 966–974.
- Houra, K.; Turčić, P.; Gabričević, M.; Weitner, T.; Konjevoda, P.; Štambuk, N. Interaction of α-Melanocortin and Its Pentapeptide Antisense LVKAT: Effects on Hepatoprotection in Male CBA Mice. Molecules 2011, 16, 7331–7343.
- Mohri, M.; Rezapoor, H. Effects of heparin, citrate, and EDTA on plasma biochemistry of sheep: Comparison with serum. Res. Vet. Sci. 2009, 86, 111–114.
- Minarova, H.; Palikova, M.; Mares, J.; Syrova, E.; Blahova, J.; Faldyna, M.; Ondrackova, P. Optimisation of the lymphocyte proliferation assay in rainbow trout (Oncorhynchus mykiss). Vet. Med. 2019, 64, 547–557.
- Root-Bernstein, R. Simultaneous origin of homochirality, the genetic code and its directionality. Bioessays 2007, 29, 689–698.
- Root-Bernstein, R. Experimental test of L- and D-amino acid binding to L- and D-codons suggests that homochirality and codon directionality emerged with the genetic code. Symmetry 2010, 2, 1180–1200.
- Turčić, P.; Bradamante, M.; Houra, K.; Štambuk, N.; Kelava, T.; Konjevoda, P.; Kazazić, S.; Vikić-Topić, D.; Pokrić, B. Effects of α-Melanocortin Enantiomers on Acetaminophen-Induced Hepatotoxicity in CBA Mice. Molecules 2009, 14, 5017–5026.
- Arquès, D.G.; Michel, C.J. A complementary circular code in the protein coding genes. J. Theor. Biol. 1996, 182, 45–58.
- Michel, C.J. The maximal C3 self-complementary trinucleotide circular code X in genes of bacteria, eukaryotes, plasmids and viruses. J. Theor. Biol. 2015, 380, 156–177.
- Bartonek, L.; Braun, D.; Zagrovic, B. Frameshifting preserves key physicochemical properties of proteins. Proc. Natl. Acad. Sci. USA 2020, 117, 5907–5912.
- Štambuk, N. On circular coding properties of gene and protein sequences. Croat. Chem. Acta 1999, 72, 999–1008.
- Štambuk, N. Universal metric properties of the genetic code. Croat. Chem. Acta 2000, 73, 1123–1139.
- Wichmann, S.; Ardern, Z. Optimality in the standard genetic code is robust with respect to comparison code sets. Biosystems 2019, 185, 104023.
- Wichmann, S.; Scherer, S.; Ardern, Z. Computational design of genes encoding completely overlapping protein domains: Influence of genetic code and taxonomic rank. bioRxiv 2020.
- Youvan, D.C. Mathematics of the Genetic Code. Available online: https://www.youvan.com/Mathematics of the Genetic Code-submit 2-redacted.pdf (accessed on 23 March 2021).
- Füllen, G.; Youvan, D.C. Genetic algorithms and recursive ensemble mutagenesis in protein engineering. Complex Int. 1994, 1. Available online: http://www.complexity.org.au/ci/vol01/fullen01/html/ (accessed on 23 March 2021).
- Arkin, A.P.; Youvan, D.C. An algorithm for protein engineering: Simulations of recursive ensemble mutagenesis. Proc. Natl. Acad. Sci. USA 1992, 89, 7811–7815.
- Dila, G.; Michel, C.J.; Thompson, J.D. Optimality of circular codes versus the genetic code after frameshift errors. Bio. Syst. 2020, 195, 104134.
- May, E.; Vouk, M.; Rosnick, D. An error-correcting code framework for genetic sequence analysis. J. Frankl. Inst. 2004, 341, 89–109.
- Thompson, J.D.; Ripp, R.; Mayer, C.; Poch, O.; Michel, C.J. Potential role of the X circular code in the regulation of gene expression. Biosystems 2021, 203, 104368.
- Seligmann, H.; Pollock, D.D. The ambush hypothesis: Hidden stop codons prevent off-frame gene reading. DNA Cell Biol. 2004, 10, 701–705.
- Blanchet, S.; Cornu, D.; Hatin, I.; Grosjean, H.; Bertin, P.; Namy, O. Deciphering the reading of the genetic code by near-cognate tRNA. Proc. Natl. Acad. Sci. USA 2018, 115, 3018–3023.
- Blanchet, S.; Cornu, D.; Argentini, M.; Namy, O. New insights into the incorporation of natural suppressor tRNAs at stop codons in Saccharomyces cerevisiae. Nucleic Acids Res. 2014, 15, 10061–10072.
- Solis, A.D. Amino acid alphabet reduction preserves fold information contained in contact interactions in proteins. Proteins 2015, 83, 2198–2216.
- Atchley, W.R.; Zhao, J.; Fernandes, A.D.; Drüke, T. Solving the protein sequence metric problem. Proc. Natl. Acad. Sci. USA 2005, 102, 6395–6400.
- Polyansky, A.A.; Zagrovic, B. Evidence of direct complementary interactions between messenger RNAs and their cognate proteins. Nucleic Acids Res. 2013, 41, 8434–8443.
- Koonin, E.V.; Novozhilov, A.S. Origin and evolution of the universal genetic code. Annu. Rev. Genet. 2017, 51, 45–62.
- Choi, J.; O’Loughlin, S.; Atkins, J.F.; Puglisi1, J.D. The energy landscape of −1 ribosomal frameshifting. Sci. Adv. 2020, 6, eaax6969.
- Rozov, A.; Westhof, E.; Yusupov, M.; Yusupova, G. The ribosome prohibits the G U wobble geometry at the first position of the codon–anticodon helix. Nucleic Acids Res. 2016, 44, 6434–6441.
- Fang, Y.; Treffers, E.E.; Li, Y.; Tas, A.; Sun, Z.; van der Meer, Y.; de Ru, A.H.; van Veelen, P.A.; Atkins, J.F.; Snijder, E.J.; et al. Efficient −2 frameshifting by mammalian ribosomes to synthesize an additional arterivirus protein. Proc. Natl. Acad. Sci. USA 2012, 109, E2920–E2928.
- Van Regenmortel, M.H.V. Structure-based reverse vaccinology failed in the case of HIV because it disregarded accepted immunological theory. Int. J. Mol. Sci. 2016, 17, 1591.
- Moxon, R.; Reche, P.A.; Rappuoli, R. Editorial: Reverse Vaccinology. Front. Immunol. 2019, 10, 2776.
- Štambuk, N.; Konjevoda, P. The temperature dependence of amino acid hydrophobicity data is related to the genetic coding algorithm for complementary (sense and antisense) peptide interactions. Data Brief. 2020, 30, 105392.
- Štambuk, N.; Konjevoda, P. Structural and functional modeling of artificial bioactive proteins. Information 2017, 8, 29.