2.1. Mild Versus Severe Disease Course: How Different Is the Immune Response?
A pronounced initial immune response and higher SARS-CoV-2 copy numbers at diagnosis usually correlate with the severity of disease [
8]. In fatal cases, the immune response itself is pathogenic. This can occur through the production of antibodies [
10] that damage tissues [
11,
12], or through inducing excessive inflammation, resulting in a cytokine storm [
13,
14]. The immunological measurements of the immune response to SARS-CoV-2 are concentrated so far on total IgG antibody levels, with some groups also reporting IgA, IgM, and neutralizing antibody levels. Only occasionally, memory B and T cell levels are also included in the assessments. This is a considerable deficit because the cellular immune capacity is an important element with which to control SARS-CoV-2. Better immunological metrics must be developed, and reliable biomarkers to diagnose earlier courses with a subsequent damaging immune response are missing as of now.
Figure 1 outlines in detail the important facets of the immune response found in mild compared to severe disease. The clinical manifestation of COVID-19 frequently shows smooth transitions between these two extreme phenotypes. It remains unknown which main determinants are responsible for a shift to a bad course. Advanced age [
15], cardiovascular disease (CVD), and obesity [
16] can also include the genetic factors that could be involved in causing severe COVID-19 [
15]. Nevertheless, a minor subset of young and middle-aged individuals also develop massive inflammatory responses and need intensive care treatment with mechanical ventilation [
14]. Several studies have tried to find out mediators which indicate a switch to a hyper-inflammatory reaction pattern [
13,
17,
18,
19,
20]. Genetic predisposition may play an important role in this context [
21]. Furthermore, late COVID immune responses are an emerging problem. Some individuals with a mild initial course of COVID-19 suffer from chronic symptoms with a duration of >2 months after the initial infection, called long COVID syndrome [
22,
23]. The symptoms resemble other postinfectious syndromes following outbreaks of Ebola [
24] and chikungunya [
25]. There is some overlap with myalgic encephalomyelitis [
26], with dysregulated autonomic nervous system components and perturbed immune parameters (
https://www.biorxiv.org/content/10.1101/2020.02.20.958249v2, accessed on 28 February 2020). The currently extensive impact of COVID-19 offers a great chance for science to improve the understanding of this long-lasting postinfection syndrome, which is probably based on a disturbed neuro-endocrino-immunological axis.
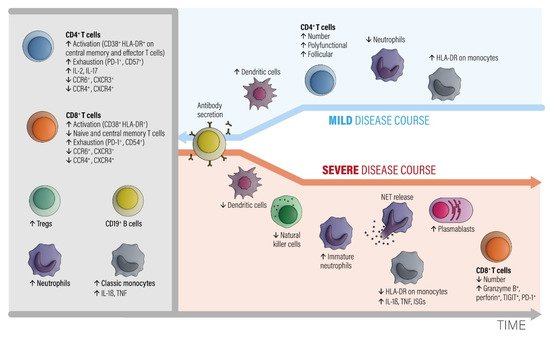
Figure 1. The involvement of different classes of immune cells mediating different courses of COVID-19 during development of the disease, modified from ref. [
27]. Abbreviations: CCR = C-C chemokine receptor. CTLA-4 = cytotoxic T-lymphocyte protein 4. CXCR = C-X-C chemokine receptor. HLA-DR = HLA DR isotype. IL = interleukin. ISGs = interferon-stimulated genes. NETs = neutrophil extracellular traps. PD-1 = programmed cell death 1. TNF = tumor necrosis factor. Tregs = regulatory T cells. Modified from [
27].
2.1.1. Grey Area
In the early phase of COVID-19, activated peripheral blood CD4+ and CD8+ T lymphocyte fractions arise in the circulation. CD4+ T cells show an increased level of IL-2 and IL-17 production. T regulatory cell (Tregs), neutrophil, and classic monocyte levels are also increased. Numbers of B cells and monocytes remain unchanged and increase later on in the course. The CD4+ and CD8+ T effector and memory lymphocytes are activated (CD38+, HLA-DR+) and show exhaustion-associated markers (PD-1, CD54). The chemokine receptor expression pattern shows decreased CCR6, CCR4, CXCR3, and CXCR4 antigens.
2.1.2. Blue Area
A subsequent favorable clinical course is associated with increased numbers of circulating dendritic cells, recovery of T cell numbers by polyfunctional and follicular helper T cell fractions, decreased neutrophils, and an increased number of intermediate HLA-DR-expressing monocytes. The humoral antibody response starts via activated and expanded B cells.
2.1.3. Red Area
Bad disease courses show decreased levels of dendritic and natural killer cell fractions and increased levels of immature neutrophils. Monocytes downregulate HLA-DR expression and upregulate IL-1β, TNF, and interferon-stimulated genes (ISGs). Although low in absolute numbers, CD8+ T cells upregulate granzyme B and perforin secretion. While the number of plasma blasts increases, specific antibodies are consistently produced. The main point is that myeloid cells show pro-inflammatory behavior with an egress of immature neutrophils from the bone marrow and an enhanced formation of neutrophil extracellular nets (NETs). NET formation is an important contributor to the development of organ and endothelial injury in conjunction with activation of the complement and coagulation cascades and a so-called cytokine storm.
2.3. How Do Innate and Adaptive Immune Responses Contribute to the Course of COVID-19?
- (A)
-
Innate immune response (recognition, interferon, and inflammasome activation)
After entering target cells via the ACE2 receptor, SARS-CoV-2 is detected by pattern-recognition receptors that resemble Toll-like receptors 3, 7, 8, and 9, as well as by the viral-infection sensors RIG-I and MDA5 [
14,
32]. The recognition stimulates the type I interferon (IFN) response and activates IFN-dependent genes [
14,
33]. The release of danger-associated molecular patterns (DAMP) contribute to the activation of the NLRP3 inflammasome [
34] and other inflammasome complexes. The NLRP3 inflammasome induces caspase-1-dependent cleavage and the release of the key proinflammatory cytokines interleukin-1ß (IL-ß) and IL-18, and this correlates with COVID-19 disease severity [
14,
35]. Furthermore, gasdermin-D-mediated pyroptotic cell death is triggered [
14,
36]. Due to cell death, lactate dehydrogenase (LDH) is produced. These data underline that inflammasome activation is an important feature of COVID-19 which is reflected by high LDH blood levels [
14,
37]. Similar to SARS-CoV and MERS-CoV, SARS-CoV-2 is able to inhibit type I IFN responses in infected cells [
38,
39,
40,
41]. This allows the virus to replicate itself and to induce stronger tissue damage, and therefore an overwhelming immune response (see
Figure 2, severe SARS-CoV-2 disease). Hence, the immune system struggles to limit viral replication while managing dying and dead cells [
14]. Immune cells begin to also flow into the lungs, where they produce high amounts of proinflammatory cytokines, a process that escalates the situation [
14]. The imbalanced immune reaction caused by the impaired type I IFN response contributes significantly to the overall severity of acute COVID-19 [
13,
41,
42,
43] by inducing a prolonged and ineffective innate immune response (
Figure 2). This is also emphasized by new results coming from the COVID Human Genetic Effort, which found that inborn errors in the type I IFN pathway [
44], or the presence of neutralizing autoantibodies to type I IFNs [
11,
45], were over-represented among individuals who developed life-threatening COVID-19. If these imbalanced or impaired innate responses also contribute to the development of MIS-C and long COVID remains to be determined [
14]. In any case, an overwhelming inflammasome activation is an important component of severe COVID-19 [
37]. This pathway also triggers a coagulation cascade which explains coagulopathy and severe thrombotic events in patients with severe COVID-19 [
36,
46].
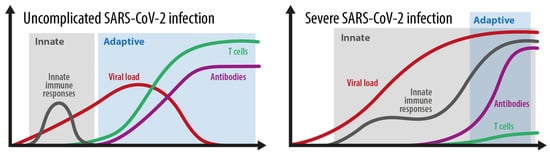
Figure 2 shows the different kinetics shown in an average and in a severe SARS-CoV-2 infection. During a simple SARS-CoV-2 infection, the innate immune response phase gains rapid control of the viral load and is replaced by an effective and adaptive immune response based on adequate antibodies and T cell control. During a severe infection, the balance between innate and adaptive immune responses is essentially disturbed. An ineffective innate immune response with a probably weak type I IFN response does not gain control over the virus, and therefore prolongs and overlaps with an ineffective adaptive immune response. An ineffective adaptive immune response is characterized by a very weak and delayed adaptive T cell response. This situation allows the virus to expand, which usually leads to an overwhelming systemic inflammatory reaction.
Figure 2. The kinetics of the innate and adaptive immune responses in simple versus severe SARS-CoV-2 infections. Grey area: innate immune response. Blue area: adaptive immune response. Adopted from [
52].
- (B)
-
Adaptive immune response (Antibody production, seroconversion, T cell memory development)
The adaptive immune responses induced by SARS-CoV-2 infection largely follow the expected patterns observed in other comparable viral infections. Thus, more than 90% of infected individuals seroconvert a few weeks after initial infection [
47,
48], and anti-spike IgG antibodies are associated with protection from reinfection. T cell responses to the SARS-CoV-2 spike protein parallel B cell responses to the similar protein in nearly all COVID-19 cases [
49] (
Figure 2). Additionally, unexposed people occasionally show T cell reactivity to SARS-CoV-2 due to cross-reactive immunity to common cold coronaviruses [
49]. The so-called antibody-dependent enhancement (ADE) has been supposed as another possible mechanism of severe COVID-19. These antibodies can facilitate Fc-receptor-mediated endocytosis of the virus and enhance viral replication resulting in massive hyperinflammation. ADE has been proved in dengue [
50] and MERS [
51] but clear evidence for its involvement in severe SARS-CoV-2 infections as well as SARS-CoV-2 second infections is lacking so far (
https://doi.org/10.1016/S1473-3099(20)30783-0, accessed on 2 May 2021). Furthermore, to what extent this putative mechanism may also influence the balance between innate and adaptive immune responses in severe COVID-19 as shown in
Figure 2 is not known so far.
2.4. How Strongly Does the Immune Response to SARS-CoV-2 Differ between Individuals?
Immune responses can vary markedly. Some people generate a very effective immune response. They cannot be infected again, and they will not give the virus to anyone else (so called ‘sterilizing’ immunity). Other people produce antibodies and are protected from disease, but not from future infection by SARS-CoV-2 [
28]. These people still pass the virus. The number of antibodies produced after infection, the quality of those antibodies (how good they are at preventing infection) and the number and quality of the T cell response generated vary markedly between individuals. It is not known in detail so far for how long and how strong this immunity is over a longer period of time. With the rapidly growing proportion of the world’s population having been infected with SARS-CoV2, the degree to which this group is protected from reinfection is increasingly of interest. The waning of neutralizing antibody responses over the first year after infection makes reinfections more possible [
53]. However, B cell and T cell memory responses induced by primary infection suggest that reinfection severity, and potentially transmission, may be mitigated over the longer term [
53]. The potential of higher levels of neutralizing antibodies to be induced by vaccination suggests that reinfection could be further reduced by vaccination of those who have previously been infected [
53]. Undoubtedly, a better understanding of all those factors, which influence immunity, is urgently needed to help achieve long-term immune control of the SARS-CoV-2 pandemic.
- (A)
-
The influence of age and lifestyle
Age is an important factor. Usually, the risk for severe COVID-19 increases abruptly above age 70 [
54,
55,
56]. Nevertheless, this may not be true for all elderly people. Some studies reported that antibody responses to infection do not vary with age in adults [
57] and were even higher in older patients [
58,
59]. This was recently confirmed in 217 participants obtained from the Austrian Ischgl cohort by a 7–8 months (follow up) after infection [
60]. Lifestyle factors associating with increased low-grade inflammation may explain these discrepancies. Preexisting obesity, hypertension, chronic obstructive pulmonary disease, cardiovascular disease (CVD) and smoking associate stronger with severe COVID-19 courses [
61]. Notably, smoking induces, besides other negative effects, the expression of angiotensin-converting enzyme 2 (ACE2), which allows SARS-CoV-2 to enter cells [
62]. Furthermore, an increased neutrophil to lymphocyte ratio (NLR) is a surrogate marker for systemic inflammation and is associated with poor prognosis in COVID-19 [
63]. The NLR increases with the degree of obesity especially in context with metabolic syndrome and type 2 diabetes [
64]. Older individuals with such conditions show two characteristics of severe COVID-19, such as failure in developing sufficient antiviral immune responses and a tendency to develop uncontrolled exacerbating responses to infections resulting in hyperinflammation and acute respiratory distress syndrome [
14]. Older individuals also have weaker type I IFN responses, which further aggravate situations [
65]. Also, additional “inflammaging” markers like NLPR3 activation [
37], IL-6, IL-12 and IL-1ß secretion [
66], and danger-related molecular patterns, including high mobility group box 1 (HMGB1) [
67] have shown to be predictive for a severe COVID-19 course in elderly. The majority of young people have mild COVID-19 disease [
68]. This is interesting because, although newborns and young children produce lower amounts of type I IFN upon stimulation through the virus and show a reduced breadth of antibody responses to SARS-CoV-2 proteins, they experience mild COVID-19 [
58,
69]. Possible explanations may be given to this by protective action taken by cross-reactive antibodies to common-cold coronaviruses and constitutive differences of the immune system [
70]. Furthermore, the immune system of young children is more accustomed to face novel challenges, while the elderly mainly rely on memory driven immune responses [
14]. This may explain the more benign clinical courses in the young.
- (B)
-
The influence of gender
Men have a much greater risk of severe COVID-19 courses but women more frequently develop long COVID [
71]. It is interesting that, in this context, women elicit stronger type I IFN responses after stimulation with TLR7 ligands [
72], that they develop stronger vaccine responses and that they have better survival rates for a number of acute infections than men [
73]. These sex differences are not age dependent and, thus, can also be seen before puberty between boys and girls, which indicates rather genetic differences. Interestingly, TLR7, a common virus sensor, is expressed on the X chromosome, suggesting a possible gene-dosage effect between the sexes [
74]. Furthermore, neutralizing autoantibodies to type I IFN are much more frequently found in male patients with COVID-19 [
14]. Future research is warranted to elucidate the reason for this phenomenon. The thymus involutes more rapidly in boys than in girls which may explain a weaker T cell control in men [
75]. Otherwise, MIS-C is quite evenly distributed between boys and girls [
76]. It is also important to which extent social factors and differing exposure play a part in sex differences [
14].
- (C)
-
The influence of immune deficiency
People with immunodeficiency, autoimmune diseases, or those who take immunosuppressive medication respond less to SARS-CoV-2 than young healthy people [
77]. However, the data are also contradictory for this. One systematic review found no statistically significant increased risk of severe COVID-19 in immunosuppressed persons [
78] whereas in cases with solid-organ transplants and cancer patients an increased risk was seen [
79]. Especially, cancer patients treated with checkpoint inhibitors may be at a high risk to develop severe COVID-19 [
80]. On the other hand, it is also possible that the host immune response against SARS-CoV-2 can cause an anticancer effect in certain constellations. Challenor et al. [
81] reported one case with classical Hodgkin lymphoma with stage III disease that went into remission without corticosteroids or immunochemotherapy. Thus, the SARS-CoV-2 triggered immune activation may mediate an anti-tumor immune response as seen in other infections in the context of high-grade non-Hodgkin lymphoma [
82]. Future research is needed to clarify the outcome of cancer patients who overcome COVID-19 disease. Particularly tumor patients with good checkpoint inhibitor effects that successfully overcome COVID-19 disease should be subject to research [
83]. Furthermore, the effects of immunosuppressive medications, especially methotrexate and rituximab, on decreasing serological responses must be determined in future studies. Concerning successful vaccination against SARS-CoV-2 in rheumatic diseases, methotrexate may be held for up to 2 weeks after the vaccination, and rituximab a few weeks after the vaccination until further clinical trials can answer this question [
84]