Human papillomaviruses (HPVs) are involved in the development of around 5% of all human cancers and HPV16 is the high-risk genotype with the highest prevalence worldwide, playing a dominant role in all HPV-associated cancers. Recombinant antibodies against specific antigens have shown great promise for the therapy of infectious diseases and cancer.
- antibody therapeutics
- recombinant antibodies
- intracellular antibodies
- single-chain antibody fragment
- nanobody
- Human papillomaviruses
- HPV oncoproteins
- HPV-associated cancer
- HPV cancer therapy
1. Introduction
In the last decades, thanks to DNA technology, recombinant antibodies have been increasingly applied to the therapy of many diseases of genetic, infectious, or tumour origin. The mammalian immune system produces antibodies against virtually any antigen, and over 30 years of molecular technology studies allowed to establish methods to select ligands for specific protein epitopes in either intra- or extra-cellular environment from libraries of different kinds, even originating from animals immunized with antigens of interest. Recombinant antibodies can interfere with specific protein functions at DNA, RNA, or protein level, and many antibodies and antibody-based products either approved or under investigation in clinical trials are revolutionizing classical drug-based tumour chemotherapy[1].
Phage display, yeast display, ribosome display, bacterial display, mammalian cell surface display, mRNA display, and DNA display technologies allow in vitro selection from non-animal-derived recombinant (naïve or synthetic) repertoires (libraries) of peptides and antibody fragments in different formats specific for chosen targets. All display technologies mimic what occurs in vivo during antibody generation by the immune system as they rely on (1) genotypic diversity (2) link between genotype and phenotype; (3) selective pressure for increasing antibody specificity; (4) amplification of specific clones originated by selective pressure. The coding sequences of the binders identified can be expressed in prokaryotic or eukaryotic systems and tested both in vitro and in vivo for their ability to recognize the target antigen and counteract its activity.
Specific antibodies in different formats representing more or less extended regions of an immunoglobulin (Ig) can be delivered directly to the cells as purified proteins or expressed as intracellular antibodies (intrabodies) by recombinant DNA technology. Antibodies in single-chain format (scFvs) and single-domain antibody or nanobodies (sdAb or Nbs)[2], are the most suitable for expression as intrabodies because of low molecular weight and easy engineering[3].
2. Antibody structure: scFvs and nanobodies
A whole IgG molecule (150 kDa) comprises heavy (H) and light (L) chains each consisting of a variable (VH and VL) and a constant (CH and CL) region covalently linked to each other and to oligosaccharides necessary for antibody effector functions and for long serum half-life. The antigen-binding regions responsible for diversity among antibodies are the complementary determining regions (CDRs), three for each VH and VL. The VH and VL joined by a disulphide bond and covalently linked to the first CH domain are obtainable by IgG proteolysis, resulting in a Fab monovalent antibody fragment (55 kDa) (Figure 1).
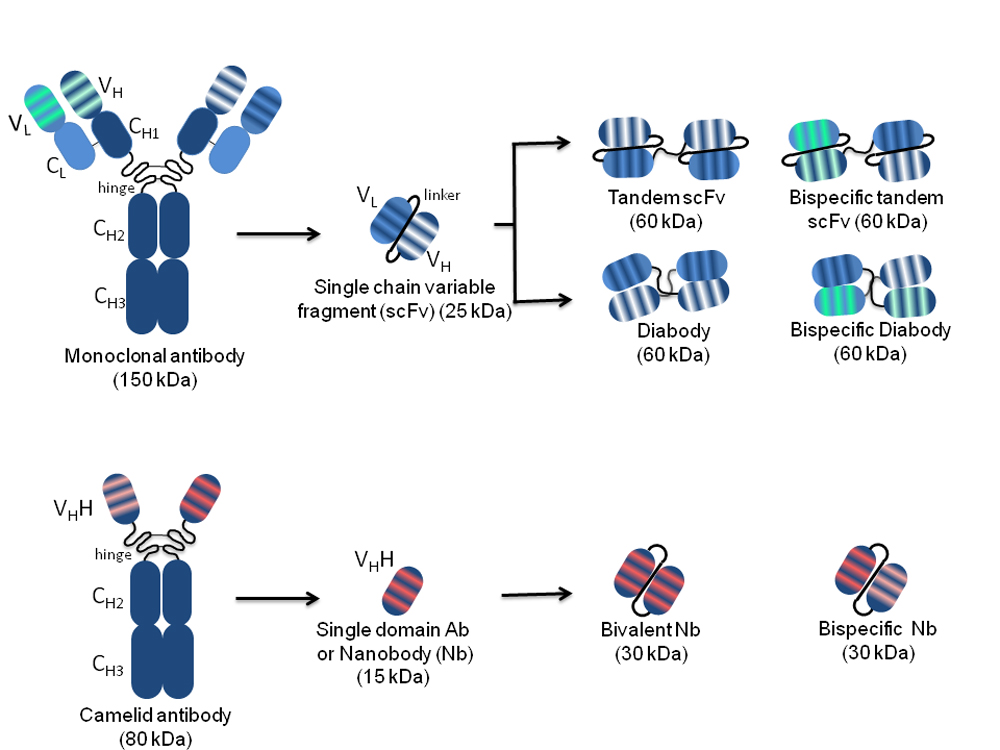
Figure 1. Schematic representation of the structure of conventional and camelidae monoclonal, and single-chain antibodies. On the left, the whole monoclonal antibody (top) and camelid antibody (bottom) structures are represented. The variable light (VL) and heavy (VH) chains, as well as the constant light (CL) and heavy (CH1, CH2, CH3) chains are indicated. The complementarity determining regions responsible for antigen binding, three for each VL and each VH, are represented by stripes highlighted in different colours according to the different antigen specificity. Arrows indicate different monospecific and bispecific antibody fragments derived from the original antibody molecules with their nomenclature and molecular weight. The VH and VL in the different arrangements are connected by peptide linkers of 3–12 amino acids represented by black curved lines.
The N-terminal IgG fragment including the VH and VL retains the same antigen-binding capacity as the whole IgG molecule. The antibodies in single-chain format, called scFvs (27 kDa), lack the constant regions and include only the VH and VL linked by a short peptide consisting of a sequence of glycine and serine residues such as (Gly4Ser)3. This arrangement provides flexibility, hydrophilicity, and resistance to proteases digestion. The linker length can be modified to favour or not the formation of multimers. A short linker (3–12 amino acids) prevents the formation of monomeric forms and supports inter-molecular VH-VL combinations with spontaneous formation of a scFv dimer called “diabody” (60 kDa), where each of the two antigen-binding sites is formed by the VH of one scFv and the VL of the other one. The linkage of two scFvs in a unique molecule forms a tandem scFv. Both diabodies and tandem scFvs can have two different binding specificities, and in this case, they are called bispecific (bs).
The CDRs of a scFv are embedded in an amino acid scaffold of either human or animal origin, according to the library utilized for selection. However, CDRs with specific binding activities can be isolated and grafted onto different scaffolds. Importantly, the use of human scaffolds allows bypassing the risk of immune reactions during clinical use.
Antibody engineering by cloning of scFv sequences into eukaryotic vectors allows the expression of scFvs as intrabodies capable of recognizing target antigens in intracellular compartments where they are located, with outcomes ranging from direct antigen blockade to indirect impairment of its activity through delocalization, to its targeted degradation[4][5]. Nbs derive from Camelidae species (e.g., llamas, dromedaries and camels), which in their antibody repertoire have IgG lacking both light chains and CH1 domains (heavy-chain-only antibodies: HCAbs)[6]. The variable domains of these HCAbs are named VHHs and can be isolated as sdAbs, which are small-sized (~15 kDa) but retain the antigen-binding capability of the full-size antibody. VHHs can easily penetrate tissues and access cryptic epitopes, are quickly cleared from the bloodstream, are more soluble and capable of efficient folding with respect to conventional mAbs, and can resist a wide pH range and high temperatures[7][8]. Despite the non-human origin, VHHs are rarely immunogenic due to the small size and high sequence homology to the human VH3 gene family, which avoids the necessity of humanization for translation into clinic[9]. Such characteristics, together with the short half-life (2 hours) facilitate application in diagnosis rather than therapy. A humanized VHH (Caplacizumab, CabliviTM) has been recently approved in Europe and USA for the treatment of acquired thrombotic thrombocytopenic purpura[10][11][12], and others targeting haematological, oncological, infectious, inflammatory/auto-immune, bone and neurological diseases are being evaluated in clinical trials.
3. HPV-Associated Lesions and Therapies
HPVs are small, non-enveloped viruses, with a circular dsDNA genome of about 8 Kbp and an icosahedral capsid composed of the major L1 protein and the minor L2 protein. Following infection of the epithelium basal cells, the E1 and E2 early proteins play key roles in viral DNA replication and amplification, as well as in regulating viral transcription. When actively expressed, the viral genome is maintained as an episome. Viral genome amplification, late gene expression, and viral progeny assembly are limited to terminally differentiated layers of the epithelium. To overcome growth arrest of terminally differentiated keratinocytes and be able to replicate, HPV genomes express E6, E7, and E5 proteins, which keep the infected cells in a competent state for DNA synthesis. Most HPV infections usually clear within 1–2 years. Nevertheless, in some cases, the virus may persist and occasional integration of its genome in the host’s genome occurs, with the interruption of the E2 transcriptional repressor coding sequence and consequent overexpression of E6 and E7. Persistent expression of these proteins leads to cancer development by impacting on cell cycle control, cell growth regulation and resistance to apoptosis (Figure 2).
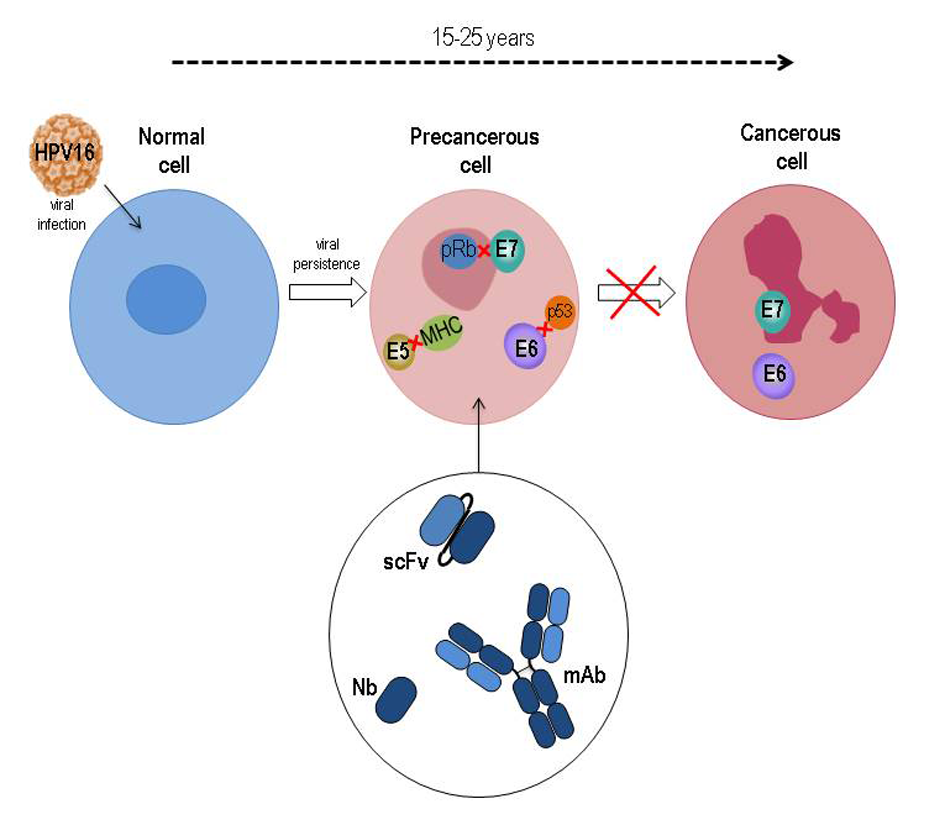
Figure 2. Schematic representation of HPV16-related cancer natural history with the underlying role of E5, E6 and E7 oncoproteins during transition from normal to precancer and cancer cell. The main targets of HPV oncoproteins are indicated. Oncoprotein-specific antibodies in different formats are potential therapeutics to counteract tumor progression.
The E7 protein binds to pRB and displaces the E2F transcription factor, thus facilitating S-phase entry[13]. E7 also alters p21, histone deacetylases (HDACs) and cyclins and alters their function. The E6 interacts with PDZ-containing proteins through a PDZ-domain binding site and thus affects cell polarity, cell-cell adhesion, and diverse cell signaling pathways[14]. Recently, its X-ray structure in complex with E6-associated protein (E6AP), the ubiquitin ligase involved in p53 degradation, and p53 has been resolved[15].
More recently, it has also been found that E5, a small hydrophobic oncoprotein mostly located in the endopalsmatic reticulum (ER) and Golgi apparatus, also plays an important role in immune evasion, cell growth and interference with signal transduction pathways, and cooperates with E6 and E7 in procarcinogenic activity[16].
HPV infections are among the most common sexually-transmitted diseases and cause annually around 5% of all cancers worldwide[17][18][19]. Some genotypes, defined as high-risk (HR) types, are responsible for morbidity and mortality related to cancer, mostly of the cervix (CC) but also of the anogenital area and of the oropharynx. Currently, treatment of HPV-associated lesions varies widely, mainly depending on lesion grade and localization, and tumour stage. The therapeutic approaches currently in use aim at eliminating abnormal/malignant cells through surgery, chemotherapy, radiotherapy, targeted therapy, or immunotherapy alone or in combination. Recombinant vaccines consisting of DNA-free virus-like particles (VLPs) containing the viral L1 protein and adjuvants, have become available since more than 10 years. Nevertheless, effective prevention of the HPV-associated pathologies is expected only in the long-term if HPV vaccination is able to reach a significant percentage of the target population worldwide[20][21][22]. Of note, the 73rd World Health Assembly, held on August 2020, strongly encouraged the acceleration of actions aimed at eliminating CC as a public health problem [23].
Since during natural infections E6- and E7-specific CD4 and CD8 immune responses are associated with the regression of HPV-associated cervical lesions, both these tumour-associated antigens represent the ideal antigens for the development of therapeutic vaccines. Also the E5 oncoprotein has recently been proposed as a possible immunotherapy target[23]. Different peptide- and protein-based therapeutic vaccines against HPV-associated tumours are under development. However, anti-HPV tumours vaccines are not yet available for clinical practice in spite of the numerous E6/E7-based clinical experimentations because the lack of specific adjuvants of cytotoxic T lymphocytes (CTLs) responses often caused a poor vaccine efficacy leading to the interruption of many clinical trials in the early stages[21][22].
4. Recombinant Antibodies Targeting HPV Oncoproteins
Safe and non-invasive therapeutic interventions, without side effects and possibly not involving the individual immune response to be effective also in immunodeficient patients, would be desirable for the treatment of HPV-related lesions.
An effective and timely treatment of pre-neoplastic lesions could avoid their progression toward invasive cancer. At the same time, a well-timed and effective therapy for already established tumours could ameliorate patient prognosis. Another important therapeutic area of intervention could be the prevention of metastases deriving from surgically removed HPV-driven tumours.
In view of their crucial role in the onset and progression of HPV-driven tumours[24], E5, E6, and E7 proteins represent ideal targets for alternative anti-tumour therapeutic approaches based on protein knock out or knock down methods. In this context, recombinant antibodies seem to represent a valid therapeutic opportunity. In recent years, such antibodies have been tested for their therapeutic potential against HPV-associated disease. They are summarized here referring to their targets (Tab. 1).
Table 1. Summary of antibodies targeting the HPV16 E6, E7 and E5 oncoproteins (in transcription order). The antibody target, format, known in vitro and in vivo activities, and delivery system are indicated for each antibody. EGFR: epidermal-growth factor receptor; ER: endoplasmic reticulum; mAb: monoclonal antibody; PAGE: polyacrylamide gel electrophoresis; scFv: single-chain variable fragment; VHH: variable domain of heavy chain antibody; Zn: zinc.
|
Target |
Name |
Format |
In vitro activity |
In vivo activity |
Delivery |
References |
HPV16 E6(N-terminus) |
1F1 | scFv | -inhibition of E6/p53 binding-inhibition of p53 degradation | [25] | ||
HPV16 E6(N-terminus) |
6F4/IF4-P41L (intracellular) | scFv | -inhibition of p53 degradation-inhibition of E6AP-dependent E6/p53 binding-no p53 accumulation in 6F4 protein-transduced HPV16+ cells-induction of apoptosis in HPV16+ cells (IF4-P41L) | -6F4: lipid-based transfection-IF4-P41L: adenoviral vector-based delivery | [25][26][27][28] | |
HPV16 E6(2nd Zn binding domain-ZD2) |
1F53B83F8 | mAb | -no inhibition of p53 degradation-inhibition of E6AP-independent E6/p53 binding-no p53 accumulation in 3F8 protein-transduced HPV16+ cells | 3F8: lipid-based transfection | [26][28] | |
HPV16 E6 |
4C6 | mAb | -inhibition of p53 degradation-in 4C6 protein-transduced HPV16+ cells:-p53 accumulation-inhibition of proliferation | lipid-based transfection | [28] | |
HPV16 E6(1st Zn finger) |
GTE6-1 | scFv | -inhibition of p53 degradation-in HPV16+ cells:-p53 rescue-apoptosis induction | transfection of recombinant plasmid | [29] | |
HPV16 E6 |
F127-6G6 | mAb | p53 rescue in HPV16+ cells | sonoporation | [30] | |
HPV16 E6 |
I7nuc | scFv intrabody | -co-localization with E6-partial p53 rescue-inhibition of cell proliferation-induction of necrosis and apoptosis-detection of endogenous E6 | -prevention of tumour onset, and-impairment of established tumour growth in C57BL/6 mice | electroporation | [31][32][33] |
HPV16 E6 |
C1P5 | mAb | impairment of tumour growth in Balb/c nude mice | intratumour/intraperitoneal injection as purified protein | [34] | |
HPV16 E6 |
26 potential E6-binding clones | VHH (nanobody) | clone 2A17: detection of recombinant E6 in native PAGE | [35] | ||
HPV16 E6 |
Nb9 | VHH (nanobody) | inhibition of HPV16+ cell proliferation | inhibition of xenograft tumours in nuce mice | -subcutaneous injection of HPV16+ cells expressing Nb9 (lentiviral vector-based expression)-in vivo transfection of established tumours with Nb9-intrabody expression plasmid | [36] |
HPV16 E7 |
TVG701Y | mAb | impairment of tumour growth in Balb/c nude mice | intratumoural/intraperitoneal injection of purified mAb | [34] | |
HPV16 E7 |
pSTCF with secretory or ER signal | scFv | -down-regulation of 16E7 expression-inhibition of HPV16+ cell proliferation | Adenovirus Poly Lysine (AdpL)-based transfection of expression vectors | [37] | |
HPV16 E7 |
scFv32scFv43/scFv43M2scFv51 | scFv | -43, 51 and 43M2SD: inhibition of HPV16+ cell proliferation-43M2SD: E7 delocalization and accumulation in ER | 43M2:-prevention of tumour onset, and-impairment of established tumour growth in C57BL/6 mice | -electroporation-exosome-based delivery | [32][38][39][40][41] |
HPV16 E7 |
Nb2 | VHH (nanobody) | -binding to endogenous E7-inhibition of HPV16+ cell proliferation | liposome-based transfection of recombinant plasmids | [42] | |
HPV16 E5 |
H2-I | scFv | -detection of endogenous E5 in W12 cells-co-localization with EGFR | [43] |
4.1. E6-Specific Recombinant Antibodies
4.1.1. ScFvs and mAbs
The 1F1 and 6F4 (F4) scFvs, derived from mAbs obtained by mice immunization with the GST-HPV16E6 fusion protein and targeting the E6 N-terminus, were able to hamper p53 degradation in vitro by inhibiting the formation of the E6/p53 complex[44]. The capacity of intracellular folding and cytosolic stability/solubility of scFvF4 was improved by mutagenesis, obtaining the IF4-P41L scFv[26]. Such scFv expressed by adenoviral system was able to cause specific apoptosis of HPV16-positive cells in a way proportional to the scFv solubility and not related to p53 rescue[27]. By comparing the activity of scFv F4 to that of three anti-16E6 mAbs (1F5, 3B8, 3F8) targeting the 16ZD2 zinc-binding domain, the mAbs resulted to be unable to affect the E6AP-dependent and able to affect the E6AP-independent binding of p53, possibly as a consequence of an antibody-induced conformational change at the E6AP-binding site of E6[26].
One anti-16E6 mAb (4C6) delivered to HPV16- and HPV18-positive cells induced a specific p53 accumulation in the nucleus of HPV16-positive cells, associated to the impairment of cell proliferation in the absence of apoptosis[27].
GTE6-1, a 16E6 binder selected from a scFv library, was able to bind to the first zinc finger of E6 with high affinity. GTE6-1 was able to recognize specifically both partially denatured and native E6 and to inhibit E6-mediated degradation of p53 in vitro assay[29]. Expression of this scFv in a number of cell lines, the proliferation of which depends on E6 and E7, induced significant p53 rescue and nuclear apoptosis in HPV16-positive cells
The delivery of the 16E6-targeting F127-6G6 mAb to HPV16-positive cells by sonoporation reduced the E6-mediated p53 degradation but did not induce apoptosis[30]. Nevertheless, the effect was transient probably due to the inability of molecules as large as mAbs to penetrate the cell nucleus, and the outcome was different in the two HPV16-positive cell lines utilized, suggesting that different treatment plans might be necessary for in vivo tumour therapy. No mAb effect was observed in HPV-negative cells, confirming the safety of a mAb-based treatment, effective only in tumour cells.
The scFv I7 binder of E6 was isolated from the SPLINT library by IACT technology. In the light of E6 activity in the cell nucleus, scFvI7 was provided with a signal for nuclear localization (NLS), and found to co-localize in cell nucleus of cells co-transfected with I7nuc and recombinant E6 plasmids. Retroviral delivery of I7nuc in HPV16-positive cells caused a partial rescue of p53, associated with the block of cell proliferation and induction of apoptosis and necrosis of tumour cells[31].
The antitumour activity of I7nuc was investigated in mouse models for HPV tumours based on the injection of HPV16-positive tumour cells in C57BL6 mice. The scFv capability to either prevent cancer development from scFv-expressing tumour cells, or to hinder cancer progression by delivery to already established tumours, was evaluated. A clear impairment of tumour growth was observed in mice injected with TC-1 tumour cells expressing I7nuc, with 60% of them completely protected from tumour onset for the 4 months of observation time[31]. Similarly, tumour growth was greatly hampered following the delivery of scFvI7nuc-expressing plasmids by electroporation to implanted tumours. Histology and immunohistochemistry showed that the antitumour activity is based on the induction of extensive cell death by apoptosis in the tumour mass[32].
In view of a possible direct use of antibodies in protein format for therapeutic purposes, the scFv I7nuc protein was purified from E. coli and delivered to HPV16-positive cell lines. Interestingly, the I7nuc protein was able to penetrate cell membrane, recognize the endogenous monomeric E6 in the cell nucleus and hamper the proliferation of cells[33].
A recent study utilized anti-16 E6 and -16 E7 mAbs in an experimental murine model based on HPV16-positive cells implanted in Balb/c nude mice. Two different doses of the anti-16E6 C1P5 and anti-16E7 TVG701Y mAbs were delivered via intraperitoneal or intratumour injections and both showed significant ability to specifically inhibit tumour growth at an extent comparable to the Cisplatin chemotherapy. The mechanism underlying the mAb activity consists of a specific effect causing complement deposition and a non-specific effect on macrophage polarization facilitating the elimination of cancer cells by opsonisation, and promoting surveillance by the immune system[34].
4.1.2. Nanobodies
Nanobodies represent the new generation of anti-HPV recombinant antibodies (Figure 1). Three VHHs binding to the recombinant E6 with nanomolar affinities were identified from two llama, immune VHH phage display libraries[35]. The capacity of the selected VHHs to bind the native E6 derived from HPV16-positive biological samples had not yet been determined at the time of the study, nor had the bound E6 epitopes been characterized.
More recently, a different 16E6-targeting nanobody was isolated and characterized[36]. Such antibody could be used to counteract HPV-induced tumours given its capacity to inhibit both the proliferation of HPV16 -positive cells in vitro and the growth of xenograft tumours in nude mice.
To neutralize the E6 ability to degrade p53. VHHs against the degradation-binding domain (DBD) of p53 were developed and shown to stabilise nuclear p53 in HeLa cells, which harbour the HPV18 genome. Nevertheless, the VHHs were unable to rescue the p53 tumour-suppressive functions. The authors hypothesized that this was due to inhibition of p53 transactivation associated with an increased cell proliferation and viability[45].
4.2. E7-Specific Recombinant Antibodies
4.2.1. ScFvs and mAbs
The first scFvs selected against 16E7 oncoprotein were constructed directly from murine spleen cells, and then provided with signals for subcellular localization by cloning[37]. When the scFv-expressing plasmids were transfected in HPV16-positive cells, the scFv with SEKDEL signal for localization in the ER was effective in decreasing E7 expression in a manner inversely related to the amount of plasmid used for cell transfection. Furthermore, stable expression of these anti-16E7 scFvs and of that with localization in the ER in particular, successfully and specifically inhibited the proliferation of 16E7-expressing tumour cells.
More recently, three different scFvs against the 16E7, selected from a Phage library of human recombinant antibodies, and provided with signals for localization in the cell nucleus or ER by cloning in eukaryotic vectors, were found to inhibit significantly the proliferation of HPV16-positive cells in vitro[38]. The half-life and thermal stability of the most reactive of the anti-16E7 scFvs, scFv43, was improved by site-directed mutagenesis and the modified scFv, namely scFv43M2, was provided with the SEKDEL signal (SD) for localization in the ER[39]. The resulting scFv43M2SD was thereafter tested for its ability to counteract the 16E7 activity. In SiHa cells, the intrabody was able to subtract E7 from the usual localization and cause it to accumulate in the ER. In addition, the scFv43M2SD intracellular expression was able to inhibit significantly and specifically the proliferation of different HPV16-positive cell lines[40]. The scFv43M2 was then tested in vivo in mouse HPV tumour models, demonstrating the ability to counteract tumour progression both when administered to tumour cells before their injection into mice and when administered to already implanted tumours[32][40].
4.2.2. Nanobodies
One of four VHHs with high affinity for 16E7 selected from llama libraries by Phage display was expressed in prokaryotic system as a protein, and its ability to bind to the recombinant E7 in vitro was confirmed in immunological assays. Furthermore, the nanobody was able to detect the endogenous E7 protein in Western blotting and, most importantly, induced a specific inhibition of the proliferation of HPV16-positive cells[42].
4.3. E5-Specific scFvs
The first and currently only scFv anti-HPV16 E5 (16E5) was developed with the purpose of investigating the E5 functions. In fact, this E5-specific scFv, when expressed in human immortalized keratinocytes carrying up to 1000 episomal copies of the HPV16 genome, was able to recognize E5 and to reveal its co-localization with EGFR[43].
5. Intracellular Delivery Methods for Recombinant Antibodies against HPV Oncoproteins
Despite the safety and benefits of what would be a recombinant antibody-based therapy for HPV-associated lesions, still a low number of Nbs, scFvs and mAbs against HPV oncoproteins have been developed, and none of them has reached the clinical stage so far. One of the reasons behind this essentially lies in the difficulty of identifying a delivery method that allows recombinant antibodies to cross biological barriers while maintaining biological activity, particularly when the targets are intracellular. Several studies are underway to address this criticality and permit translation to humans. In general, recombinant antibodies for intracellular targets can be either expressed within cells from DNA plasmids or delivered directly to cells as purified proteins. This is achievable by physical methods, transfection, electroporation (EP), or fusion with a peptide transduction domain (PTD) or nanocarriers.
5.1. Electrotransfer/Electroporation
EP applies voltage pulses to generate an electric field between two electrodes, which interrupts the integrity of cell membranes with the formation of pores allowing cell uptake of nucleic acids as well as proteins. EP is therefore a safe method for intracellular protein expression since it avoids insertional mutagenesis and immunogenicity problems inherent in other methods[32][46]. The methodology is used in several, particularly immunotherapeutic, applications, and even to deliver scFvs as proteins or mRNAs[47].
5.2. Fusion with Protein Transduction Domain
PTDs or cell penetrating peptides (CPPs) are cationic and/or hydrophobic 10–30 amino acid long peptides that can be conjugated or fused to antibodies to make them able to penetrate the cell membrane via different mechanisms[48]. However, for effective translation in the clinic, the CPP-based delivery has some limitations to circumvent, mainly due to low in vivo stability and reduced binding capability.
5.3. Exosome-Based Methods
The established proof-of-concept that the Extracellular Vescicles (EV)-mediated delivery of scFvs can target intracellular antigens renders it feasible its application in vivo[41]. In addition, the possibility to obtain recombinant exosomes from the host following the administration of a genetic construct as a vaccine, suggests that delivery of anti-E6 and E7 intrabodies by exosomes is feasible in humans[49]. This also takes advantage of the capacity of the recipient organism to produce the exosomes as the intramuscular injection of DNA plasmids expressing antibody constructs, followed or not by electroporation, would permit the exosome-loaded antibodies to reach several body districts. In virtue of the antibody specificity for the HPV oncoproteins, such broad distribution will not result in off-target effects while potentially affecting any metastatic cells derived from the primary tumour.
5.4. Viral Vector-Based Methods
In the last 30 years, several clinical trials used viral vectors for gene transfer. Gamma-retroviral and lentiviral vectors for haematological cancers; adenoviral vectors for prostate, ovarian and bladder cancer; and adenovirus-associated vectors for pathologies other than cancer were employed with more or less success, and are still the object of preclinical and clinical proof-of-concept studies[50]. Noteworthy, HPV-associated lesions have a confined localization that renders them accessible to topical therapy whatever the delivery system chosen. Furthermore, the expression of the target oncoproteins being limited to cancer cells represents an additional advantage for the safety of a therapy designed to inhibit protein–protein interactions such as that based on recombinant antibodies.
5.5. Ultrasound-Based Methods
The Ultrasound-mediated targeted delivery (UMTD) is a non-invasive method that is attracting increasing interest for many biochemical applications including immunotherapy of tumours. UMTD combined with microbubbles allows delivery of therapeutic molecules precisely in the tumour site. In fact, oscillation and cavitation of microbubbles under the influence of the acoustic beam causes the reversible formation of localized pores of about 100 nm in diameter in the cell membrane[51]. This phenomenon, known as sonoporation, allows the passive release of therapeutic molecules into target cells. Feasibility and specificity of sonoporation for anti-16E6 mAb delivery to cervical carcinoma cell lines were assessed in vitro but the effect was transient and incomplete as p53 levels were affected but no apoptosis was induced[30]. However, the issue of delivery to nucleus, which probably underlies the observed partial efficacy, could be addressed using smaller antibody formats provided with NLS. Sonoporation is increasingly explored for both passive and active immunotherapy in vivo[52]. Ultrasound in combination with microbubbles even allowed the Herceptin mAb (trastuzumab) to cross the blood-brain barrier in mice, thus opening up the possibility of treating brain metastases of breast cancer[53]. However, translation of the methodology to human therapy requires further investigation on the possible elicitation of immune response by microbubbles, the exact mechanism of the therapeutic material release, the size-based microbubble capacity of penetrating cell membranes, and the excretion of microbubbles from the body.
6. Conclusions and Perspectives
Currently, recombinant antibodies for targeting antigens involved in the pathogenesis of a variety of diseases are obtainable by robust methodologies of immunization and in vitro screening. Nevertheless, their use as therapeutics may require optimization of crucial characteristics such as binding specificity and affinity, solubility, and pharmacokinetics, as well as setting up an appropriate delivery system. The possibility of designing bispecific antibodies that combine binding domains from different parental antibodies can expand the binding capacity of a single molecule by targeting at the same time multiple antigens such as E6 and E7, or multiple epitopes on the same antigen (such as DBD and E6AP binding domain on E6) but their solubility and stability may be affected and require corrections[54].
In conclusion, the therapy for HPV-associated lesions relying on antibodies shows great potential and presents some advantages over more conventional systems of immunization, such as specificity and effectiveness also in subjects immunosuppressed by natural or induced causes as co-infections or pharmacological treatments.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22179143
References
- Antibody Therapeutics Approved or in Regulatory Review in the EU or US . The Antobody Society. Retrieved 2021-10-8
- Ruei-Min Lu; Yu-Chyi Hwang; I-Ju Liu; Chi-Chiu Lee; Han-Zen Tsai; Hsin-Jung Li; Han-Chung Wu; Development of therapeutic antibodies for the treatment of diseases. Journal of Biomedical Science 2020, 27, 1-30, 10.1186/s12929-019-0592-z.
- Hélène Kaplon; Janice M. Reichert; Antibodies to watch in 2021. mAbs 2021, 13, 1860476, 10.1080/19420862.2020.1860476.
- M Visintin; The intracellular antibody capture technology: towards the high-throughput selection of functional intracellular antibodies for target validation. Methods 2004, 34, 200-214, 10.1016/j.ymeth.2004.04.008.
- Congcong Zhang; Rina M. Ötjengerdes; Julian Roewe; Rebeca Mejias-Estevez; Andrea L. J. Marschall; Applying Antibodies Inside Cells: Principles and Recent Advances in Neurobiology, Virology and Oncology. BioDrugs 2020, 34, 435-462, 10.1007/s40259-020-00419-w.
- C. Hamers-Casterman; T. Atarhouch; Serge Muyldermans; G. Robinson; C. Hammers; E. Bajyana Songa; N. Bendahman; Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446-448, 10.1038/363446a0.
- M. M. Harmsen; H. J. De Haard; Properties, production, and applications of camelid single-domain antibody fragments. Applied Microbiology and Biotechnology 2007, 77, 13-22, 10.1007/s00253-007-1142-2.
- Sophie Steeland; Roosmarijn E. Vandenbroucke; Claude Libert; Nanobodies as therapeutics: big opportunities for small antibodies. Drug Discovery Today 2016, 21, 1076-1113, 10.1016/j.drudis.2016.04.003.
- Chloé Ackaert; Natalia Smiejkowska; Catarina Xavier; Yann G. J. Sterckx; Sofie Denies; Benoit Stijlemans; Yvon Elkrim; Nick Devoogdt; Vicky Caveliers; Tony Lahoutte; et al. Immunogenicity Risk Profile of Nanobodies. Frontiers in Immunology 2021, 12, 632687, 10.3389/fimmu.2021.632687.
- Timothée Chanier; Patrick Chames; Nanobody Engineering: Toward Next Generation Immunotherapies and Immunoimaging of Cancer. Antibodies 2019, 8, 13, 10.3390/antib8010013.
- Sean Duggan; Caplacizumab: First Global Approval. Drugs 2018, 78, 1639-1642, 10.1007/s40265-018-0989-0.
- Sean Duggan; Correction to: Caplacizumab: First Global Approval. Drugs 2018, 78, 1955-1955, 10.1007/s40265-018-1031-2.
- William K. Songock; Seong-Man Kim; Jason M. Bodily; The human papillomavirus E7 oncoprotein as a regulator of transcription. Virus Research 2016, 231, 56-75, 10.1016/j.virusres.2016.10.017.
- Vats A., Trejo-Cerro O. Thomas M., Banks L; Human papillomavirus E6 and E7: What remains? . Tumour Virus Res 2021, 11, 200213, 10.1016/j.tvr.2021.200213.
- Denise Martinez-Zapien; F. Xavier Ruiz; Juline Poirson; André Mitschler; Juan Ramirez; Anne Forster; Alexandra Cousido-Siah; Murielle Masson; Scott Vande Pol; Alberto Podjarny; et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 2016, 529, 541-545, 10.1038/nature16481.
- Francesca Paolini; Gianfranca Curzio; Marcelo Nazario Cordeiro; Silvia Massa; Luciano Mariani; Fulvia Pimpinelli; Antonio Carlos de Freitas; Rosella Franconi; Aldo Venuti; HPV 16 E5 oncoprotein is expressed in early stage carcinogenesis and can be a target of immunotherapy. Human Vaccines & Immunotherapeutics 2016, 13, 291-297, 10.1080/21645515.2017.1264777.
- Catherine De Martel; Martyn Plummer; Jerome Vignat; Silvia Franceschi; Worldwide burden of cancer attributable to HPV by site, country and HPV type. International Journal of Cancer 2017, 141, 664-670, 10.1002/ijc.30716.
- Silvia de Sanjosé; Maria Brotons; Miguel Angel Pavon; The natural history of human papillomavirus infection. Best Practice & Research Clinical Obstetrics & Gynaecology 2018, 47, 2-13, 10.1016/j.bpobgyn.2017.08.015.
- Catherine de Martel; Damien Georges; Freddie Bray; Jacques Ferlay; Gary M Clifford; Global burden of cancer attributable to infections in 2018: a worldwide incidence analysis. The Lancet Global Health 2019, 8, e180-e190, 10.1016/s2214-109x(19)30488-7.
- Emily A. Burger; Allison Portnoy; Nicole G. Campos; Stephen Sy; Catherine Regan; Jane J. Kim; Choosing the optimal HPV vaccine: The health impact and economic value of the nonavalent and bivalent HPV vaccines in 48 Gavi‐eligible countries. International Journal of Cancer 2020, 148, 932-940, 10.1002/ijc.33233.
- Anna Rosa Garbuglia; Daniele Lapa; Catia Sias; Maria Rosaria Capobianchi; Paola Del Porto; The Use of Both Therapeutic and Prophylactic Vaccines in the Therapy of Papillomavirus Disease. Frontiers in Immunology 2020, 11, 188, 10.3389/fimmu.2020.00188.
- Claire Smalley Rumfield; Nicholas Roller; Samuel Troy Pellom; Jeffrey Schlom; Caroline Jochems; Therapeutic Vaccines for HPV-Associated Malignancies. ImmunoTargets and Therapy 2020, ume 9, 167-200, 10.2147/itt.s273327.
- Antonio Carlos De Freitas; Talita Helena Araújo De Oliveira; Marconi Rego Barros; Aldo Venuti; hrHPV E5 oncoprotein: immune evasion and related immunotherapies. Journal of Experimental & Clinical Cancer Research 2017, 36, 1-15, 10.1186/s13046-017-0541-1.
- Diogo Estêvão; Natália Rios Costa; Rui M. Gil da Costa; Rui Medeiros; Hallmarks of HPV carcinogenesis: The role of E6, E7 and E5 oncoproteins in cellular malignancy. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2019, 1862, 153-162, 10.1016/j.bbagrm.2019.01.001.
- Christine Giovane; Gilles Travé; Amélie Briones; Yves Lutz; Bohdan Wasylyk; Etienne Weiss; Targetting of the N-terminal domain of the human papillomavirus type 16 E6 oncoprotein with monomeric scFvs blocks the E6-mediated degradation of cellular p53. Journal of Molecular Recognition 1999, 12, 141-152, 10.1002/(sici)1099-1352(199903/04)12:2<141::aid-jmr453>3.0.co;2-o.
- Magali Lagrange; Sebastian Charbonnier; Georges Orfanoudakis; Philip Robinson; Katia Zanier; Murielle Masson; Yves Lutz; Gilles Trave; Etienne Weiss; François DeRyckere; et al. Binding of human papillomavirus 16 E6 to p53 and E6AP is impaired by monoclonal antibodies directed against the second zinc-binding domain of E6. Journal of General Virology 2005, 86, 1001-1007, 10.1099/vir.0.80607-0.
- Magali Lagrange; Charlotte Boulade-Ladame; Laurent Mailly; Etienne Weiss; Georges Orfanoudakis; François Deryckere; Intracellular scFvs against the viral E6 oncoprotein provoke apoptosis in human papillomavirus-positive cancer cells. Biochemical and Biophysical Research Communications 2007, 361, 487-492, 10.1016/j.bbrc.2007.07.040.
- Jérôme Courtête; Annie-Paule Sibler; Gabrielle Zeder-Lutz; Deniz Dalkara; Mustapha Oulad-Abdelghani; Guy Zuber; Etienne Weiss; Suppression of cervical carcinoma cell growth by intracytoplasmic codelivery of anti-oncoprotein E6 antibody and small interfering RNA. Molecular Cancer Therapeutics 2007, 6, 1728-1735, 10.1158/1535-7163.mct-06-0808.
- Heather Griffin; Robert Elston; Deborah Jackson; Keith Ansell; Michael Coleman; Greg Winter; John Doorbar; Inhibition of Papillomavirus Protein Function in Cervical Cancer Cells by Intrabody Targeting. Journal of Molecular Biology 2006, 355, 360-378, 10.1016/j.jmb.2005.10.077.
- Melissa Togtema; Samuel Pichardo; Robert Jackson; Paul F. Lambert; Laura Curiel; Ingeborg Zehbe; Sonoporation Delivery of Monoclonal Antibodies against Human Papillomavirus 16 E6 Restores p53 Expression in Transformed Cervical Keratinocytes. PLoS ONE 2012, 7, e50730, 10.1371/journal.pone.0050730.
- Carla Amici; Michela Visintin; Francesca Verachi; Francesca Paolini; Zulema Percario; Paola Di Bonito; Angela Mandarino; Elisabetta Affabris; Aldo Venuti; Luisa Accardi; et al. A novel intracellular antibody against the E6 oncoprotein impairs growth of human papillomavirus 16-positive tumor cells in mouse models. Oncotarget 2016, 7, 15539-15553, 10.18632/oncotarget.6925.
- Francesca Paolini; Carla Amici; Mariantonia Carosi; Claudia Bonomo; Paola Di Bonito; Aldo Venuti; Luisa Accardi; Intrabodies targeting human papillomavirus 16 E6 and E7 oncoproteins for therapy of established HPV-associated tumors. Journal of Experimental & Clinical Cancer Research 2021, 40, 1-11, 10.1186/s13046-021-01841-w.
- F. Verachi; Z. Percario; Paola Di Bonito; Elisabetta Affabris; C. Amici; L. Accardi; Purification and Characterization of Antibodies in Single-Chain Format against the E6 Oncoprotein of Human Papillomavirus Type 16. BioMed Research International 2018, 2018, 1-9, 10.1155/2018/6583852.
- Zewei Jiang; Joseph Albanese; Joshua Kesterson; Joshua Warrick; Rouzan Karabakhtsian; Ekaterina Dadachova; Rébécca Phaëton; Monoclonal Antibodies Against Human Papillomavirus E6 and E7 Oncoproteins Inhibit Tumor Growth in Experimental Cervical Cancer. Translational Oncology 2019, 12, 1289-1295, 10.1016/j.tranon.2019.06.003.
- Melissa Togtema; Greg Hussack; Guillem Dayer; Megan R. Teghtmeyer; Shalini Raphael; Jamshid Tanha; Ingeborg Zehbe; Single-Domain Antibodies Represent Novel Alternatives to Monoclonal Antibodies as Targeting Agents against the Human Papillomavirus 16 E6 Protein. International Journal of Molecular Sciences 2019, 20, 2088, 10.3390/ijms20092088.
- Wei Zhang; Haitao Shan; Kunpeng Jiang; Wenbin Huang; Shufeng Li; A novel intracellular nanobody against HPV16 E6 oncoprotein. Clinical Immunology 2021, 225, 108684, 10.1016/j.clim.2021.108684.
- F Wang-Johanning; G Y Gillespie; J Grim; C Rancourt; R D Alvarez; G P Siegal; D T Curiel; Intracellular expression of a single-chain antibody directed against human papillomavirus type 16 E7 oncoprotein achieves targeted antineoplastic effects.. Cancer Research 1998, 58, 1893–1900, .
- Luisa Accardi; M. Gabriella Donà; Paola Di Bonito; Colomba Giorgi; Intracellular anti-E7 human antibodies in single-chain format inhibit proliferation of HPV16-positive cervical carcinoma cells. International Journal of Cancer 2005, 116, 564-570, 10.1002/ijc.21052.
- Maria Gabriella Donà; Colomba Giorgi; Luisa Accardi; Characterization of antibodies in single-chain format against the E7 oncoprotein of the Human papillomavirus type 16 and their improvement by mutagenesis. BMC Cancer 2007, 7, 25-25, 10.1186/1471-2407-7-25.
- Luisa Accardi; Francesca Paolini; Angela Mandarino; Zulema Percario; Paola Di Bonito; Valentina Di Carlo; Elisabetta Affabris; Colomba Giorgi; Carla Amici; Aldo Venuti; et al. In vivoantitumor effect of an intracellular single-chain antibody fragment against the E7 oncoprotein of human papillomavirus 16. International Journal of Cancer 2013, 134, 2742-2747, 10.1002/ijc.28604.
- Flavia Ferrantelli; Claudia Arenaccio; Francesco Manfredi; Eleonora Olivetta; Chiara Chiozzini; Patrizia Leone; Zulema Percario; Alessandro Ascione; Michela Flego; Paola Di Bonito; et al. The Intracellular Delivery Of Anti-HPV16 E7 scFvs Through Engineered Extracellular Vesicles Inhibits The Proliferation Of HPV-Infected Cells. International Journal of Nanomedicine 2019, ume 14, 8755-8768, 10.2147/ijn.s209366.
- Shufeng Li; Wei Zhang; Kunpeng Jiang; Haitao Shan; Minke Shi; Baojun Chen; Zichun Hua; Nanobody against the E7 oncoprotein of human papillomavirus 16. Molecular Immunology 2019, 109, 12-19, 10.1016/j.molimm.2019.02.022.
- César Monjarás-Ávila; Sofía Bernal-Silva; Horacio Bach; Development of Novel Single-Chain Antibodies against the Hydrophobic HPV-16 E5 Protein. BioMed Research International 2018, 2018, 1-7, 10.1155/2018/5809028.
- Christine Giovane; Gilles Travé; Amélie Briones; Yves Lutz; Bohdan Wasylyk; Etienne Weiss; Targetting of the N-terminal domain of the human papillomavirus type 16 E6 oncoprotein with monomeric scFvs blocks the E6-mediated degradation of cellular p53. Journal of Molecular Recognition 1999, 12, 141-152, 10.1002/(sici)1099-1352(199903/04)12:2<141::aid-jmr453>3.3.co;2-f.
- Marta Celegato; Lorenzo Messa; Laura Goracci; Beatrice Mercorelli; Chiara Bertagnin; Francesca Spyrakis; Irina Suarez; Alexandra Cousido-Siah; Gilles Travé; Lawrence Banks; et al. A novel small-molecule inhibitor of the human papillomavirus E6-p53 interaction that reactivates p53 function and blocks cancer cells growth. Cancer Letters 2019, 470, 115-125, 10.1016/j.canlet.2019.10.046.
- Meijerink, M.R.; Scheffer, H.J.; Naranayan, G. Future Perspectives of IRE in Irreversible Electroporation in Clinical Practice; Meijerink, M.R.; Scheffer, H.J.; Naranayan, G, Eds.; Springer International Publishing: New York, NY, USA, 2017; pp. 271-280.
- Viggo Van Tendeloo; Peter Ponsaerts; Filip Lardon; Griet Nijs; Marc Lenjou; Christine Van Broeckhoven; Dirk R. Van Bockstaele; Zwi N. Berneman; Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood 2001, 98, 49-56, 10.1182/blood.v98.1.49.
- Kimia Kardani; Alireza Milani; Samaneh H. Shabani; Azam Bolhassani; Cell penetrating peptides: the potent multi-cargo intracellular carriers. Expert Opinion on Drug Delivery 2019, 16, 1227-1258, 10.1080/17425247.2019.1676720.
- Paola Di Bonito; Chiara Chiozzini; Claudia Arenaccio; Simona Anticoli; Francesco Manfredi; Eleonora Olivetta; Flavia Ferrantelli; Emiliana Falcone; Anna Ruggieri; Maurizio Federico; et al. Antitumor HPV E7-specific CTL activity elicited by in vivo engineered exosomes produced through DNA inoculation. International Journal of Nanomedicine 2017, ume 12, 4579-4591, 10.2147/ijn.s131309.
- A M Keeler; M K Elmallah; T R Flotte; Gene Therapy 2017: Progress and Future Directions. Clinical and Translational Science 2017, 10, 242-248, 10.1111/cts.12466.
- Yuhang Tian; Zhao Liu; Haoyan Tan; Jiahui Hou; Xin Wen; Fan Yang; Wen Cheng; New Aspects of Ultrasound-Mediated Targeted Delivery and Therapy for Cancer. International Journal of Nanomedicine 2020, ume 15, 401-418, 10.2147/ijn.s201208.
- Heleen Dewitte; Sandra Van Lint; Carlo Heirman; Kris Thielemans; Stefaan De Smedt; Karine Breckpot; Ine Lentacker; The potential of antigen and TriMix sonoporation using mRNA-loaded microbubbles for ultrasound-triggered cancer immunotherapy. Journal of Controlled Release 2014, 194, 28-36, 10.1016/j.jconrel.2014.08.011.
- Manabu Kinoshita; Nathan McDannold; Ferenc A. Jolesz; Kullervo Hynynen; Noninvasive localized delivery of Herceptin to the mouse brain by MRI-guided focused ultrasound-induced blood-brain barrier disruption. Proceedings of the National Academy of Sciences 2006, 103, 11719-11723, 10.1073/pnas.0604318103.
- Shixue Chen; Lingling Li; Fan Zhang; Yu Wang; Yi Hu; Lei Zhao; Immunoglobulin Gamma-Like Therapeutic Bispecific Antibody Formats for Tumor Therapy. Journal of Immunology Research 2019, 2019, 1-13, 10.1155/2019/4516041.