Lipoprotein(a) (Lp(a)) is a low density lipoprotein particle that is associated with poor cardiovascular prognosis due to pro-atherogenic, pro-thrombotic, pro-inflammatory and pro-oxidative properties. Traditional lipid-lowering therapy does not provide a sufficient Lp(a) reduction. For PCSK9 inhibitors a small reduction of Lp(a) levels could be shown, which was associated with a reduction in cardiovascular events, independently of the effect on LDL cholesterol. Another option is inclisiran, for which no outcome data are available yet. Lipoprotein apheresis acutely and in the long run decreases Lp(a) levels and effectively improves cardiovascular prognosis in high-risk patients who cannot be satisfactorily treated with drugs. New drugs inhibiting the synthesis of apolipoprotein(a) (an antisense oligonucleotide (Pelacarsen) and two siRNA drugs) are studied. Unlike LDL-cholesterol, for Lp(a) no target value has been defined up to now.
- lipoprotein(a)
- proprotein convertase subtilisin/kexin type 9 inhibitors
- inclisiran
- lipoprotein apheresis
1. Introduction
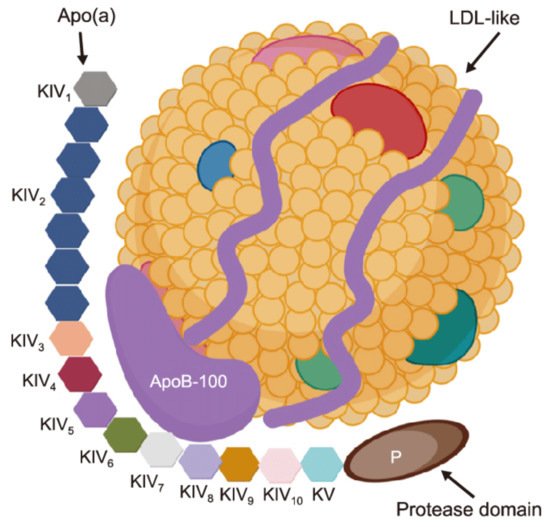
2. Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitors (PCSK9i)
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines9091271
References
- Tsimikas, S. A test in context: Lipoprotein(a): Diagnosis, prognosis, controversies, and emerging therapies. J. Am. Coll. Cardiol. 2017, 69, 692–711.
- Liu, T.; Yoon, W.-S.; Lee, S.-R. Recent Updates of Lipoprotein(a) and Cardiovascular Disease. Chonnam Med J. 2021, 57, 36–43.
- Ward, N.C.; Kostner, K.M.; Sullivan, D.R.; Nestel, P.; Watts, G.F. Molecular, Population, and Clinical Aspects of Lipoprotein(a): A Bridge Too Far? J. Clin. Med. 2019, 8, 2073.
- Lobentanz, E.M.; Krasznai, K.; Gruber, A.; Brunner, C.; Müller, H.J.; Sattler, J.; Kraft, H.G.; Utermann, G.; Dieplinger, H. Intracellular metabolism of human apolipoprotein(a) in stably transfected Hep G2 cells. Biochemistry 1998, 37, 5417–5425.
- Brunner, C.; Lobentanz, E.M.; Pethö-Schramm, A.; Ernst, A.; Kang, C.; Dieplinger, H.; Müller, H.J.; Utermann, G. The number of identical kringle IV repeats in apolipoprotein(a) affects its processing and secretion by HepG2 cells. J. Biol. Chem. 1996, 271, 32403–32410.
- Gencer, B.; Kronenberg, F.; Stroes, E.S.; Mach, F. Lipoprotein(a): The revenant. Eur. Heart J. 2017, 38, 1553–1560.
- Burgess, S.; Ference, B.A.; Staley, J.R.; Freitag, D.F.; Mason, A.M.; Nielsen, S.F.; Willeit, P.; Young, R.; Surendran, P.; Karthikeyan, S.; et al. Association of LPA variants with risk of coronary disease and the implications for lipoprotein(a)- lowering therapies: A Mendelian randomization analysis. JAMA Cardiol. 2018, 3, 619–627.
- Nordestgaard, B.G.; Chapman, M.J.; Ray, K.; Borén, J.; Andreotti, F.; Watts, G.; Ginsberg, H.; Amarenco, P.; Alberico Catapano, A.; Descamps, O.; et al. Lipoprotein(a) as a cardiovascular risk factor: Current status. Eur. Heart J. 2010, 31, 2844–2853.
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Tommasino, J.F.; Daniel, E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2019, 73, e285–e350.
- Langslet, G.; Emery, M.; Wasserman, S.M. Evolocumab (AMG 145) for primary hypercholesterolemia. Expert Rev. Cardiovasc. Ther. 2015, 13, 477–488.
- Farnier, M. An evaluation of alirocumab for the treatment of hypercholesterolemia. Expert Rev. Cardiovasc. Ther. 2015, 13, 1307–1323.
- Warden, B.A.; Minnier, J.; Watts, G.F.; Fazio, S.; Shapiro, M.D. Impact of PCSK9 inhibitors on plasma lipoprotein(a) concentrations with or without a background of niacin therapy. J. Clin. Lipidol. 2019, 13, 580–585.
- Sabatine, M.S.; Giugliano, R.P.; Wiviott, S.D.; Raal, F.J.; Dirk, J.; Blom, D.J.; Robinson, J.; Ballantyne, C.M.; Somaratne, R.; Legg, J.; et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 2015, 372, 1500–1509.
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; Stroes, E.S.; Langslet, G.; Raal, F.J.; Shahawy, M.E.; et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 2015, 372, 1489–1499.
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Huei Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. J. Med. 2017, 376, 1713–1722.
- Ridker, P.M.; Revkin, J.; Amarenco, P.; Brunell, R.; Madelyn Curto, M.; Fernando Civeira, F.; Flather, M.; Robert, J.; Glynn, R.J.; Jean Gregoire, J.; et al. Cardiovascular efficacy and safety of bococizumab in high risk patients. N. Engl. J. Med. 2017, 376, 1527–1539.
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Corinne Hanotin, C.; Harrington, R.A.; et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N. Engl. J. Med. 2018, 379, 2097–2107.
- Bittner, V.A.; Szarek, M.; Aylward, P.E.; Bhatt, D.L.; Diaz, R.; Jay, M.; Edelberg, J.M.; Fras, Z.; Goodman, S.G.; Halvorsen, S.; et al. Effect of Alirocumab on Lipoprotein(a) and Cardiovascular Risk After Acute Coronary Syndrome. J. Am. Coll. Cardiol. 2020, 75, 133–144.
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.G.; Kanevsky, E.; Gouni-Berthold, I.; Im, K.; Pineda, A.L.; Scott, M.; Wasserman, S.M.; et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Circulation 2019, 139, 1483–1492.
- Warden, B.A.; Miles, J.R.; Oleaga, C.; Ganda, O.P.; Duell, P.B.; Purnell, J.Q.; Michael, D.; Shapiro, M.D.; Fazio, S. Unusual responses to PCSK9 inhibitors in clinical cohort utilizing a structured follow-up protocol. Am. J. Prev. Cardiol. 2020, 1, 1–6.
- Edminston, J.B.; Brooks, N.; Tavori, H.; Minnier, J.; Duell, B.; Purnell, J.Q.; Kaufman, T.; Wojcik, C.; Szilard Voros, S.; Fazio, S.; et al. Discordant response of low-density lipoprotein cholesterol and lipoprotein (a) levels to monoclonal antibodies targeting proprotein convertase subtilisin /kexin type 9. J. Clin. Lipidol. 2017, 11, 667–673.
- Shapiro, M.D.; Minnnier, J.; Tavori, H.; Kassahun, H.; Flower, A.; Somaratne, R.; Fazio, S. Relationship between low-density lipoprotein cholesterol and lipoprotein (a) lowering in response to PSK9 inhibition with evolocumab. J. Am. Heart Assoc. 2019, 8, e010932.
- Raal, F.J.; Giugliano, R.P.; Sabatine, M.S.; Koren, M.J.; Blom, D.; Seidah, N.G.; Honarpour, N.; Lira, A.; Xue, A.; Chiruvolu, P.; et al. PCSK9 inhibition-mediated reduction in Lp(a) with evolocumab: An analysis of 10 clinical trials and the LDL receptor’s role. J. Lipid Res. 2016, 57, 1086–1096.
- Hoover-Plow, J.; Huang, M. Lipoprotein(a) metabolism: Potential sites for therapeutic targets. Metabolism 2013, 62, 479–491.
- Watts, G.F.; Chan, D.C.; Somaratne, R.; Wasserman, S.M.; Scott, R.; Marcovina, S.M.; Barrett, P.H.R. Controlled study of the effect of proprotein convertase subtilisin-kexin type 9 inhibition with evolocumab on lipoprotein(a) particle kinetics. Eur. Heart, J. 2018, 39, 2577–2585.
- Kostner, K.M.; Kostner, G.M. Lipoprotein (a): A historical appraisal. J. Lipid Res. 2017, 58, 1–14.
- Villard, E.F.; Thedrez, A.; Blankenstein, J.; Croyal, M.; Tran, T.-T.T.; Poirier, B.; Le Bail, J.-C.; Illiano, S.; Nobécourt, E.; Krempf, M.; et al. PCSK9 modulates the secretion but not the cellular uptake of lipoprotein(a) ex vivo: An effect blunted by alirocumab. JACC Basic Transl. Sci. 2016, 1, 419–427.
- Rader, D.J.; Mann, W.A.; Cain, W.; Kraft, H.G.; Usher, D.; Zech, L.A.; Hoeg, J.H.; Davignon, J.; Lupien, P.; Grossman, M.; et al. The low density lipoprotein receptor is not required for normal catabolism of Lp(a) in humans. J. Clin. Investig. 1995, 95, 1403–1408.
- Boffa, M.B.; Koschinsky, M.L. Update on lipoprotein(a) as a cardiovascular risk factor and mediator. Curr. Atheroscler. Rep. 2013, 15, 360.
- Stein, E.A.; Honarpour, N.; Wasserman, S.M.; Xu, F.; Scott, R.; Raal, F.J. Effect of the proprotein convertase subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia. Circulation 2013, 128, 2113–2120.
- Saavedra, Y.G.; Dufour, R.; Davignon, J.; Baass, A. PCSK9 R46L, lower LDL, and cardiovascular disease risk in familial hypercholesterolemia: A cross-sectional cohort study. Arter. Thromb. Vasc. Biol. 2014, 34, 2700–2705.
- Saavedra, Y.G.L.; Dufour, R.; Baass, A. Familial hypercholesterolemia: PCSK9 InsLEU genetic variant and prediabetes/diabetes risk. J. Clin. Lipidol. 2015, 9, 786–793.e1.
- Shapiro, M.D.; Tavori, H.; Fazio, S. PCSK9: From basic science discoveries to clinical trials. Circ. Res. 2018, 122, 1420–1438.
- Warden, B.A.; Fazio, S.; Shapiro, M.D. The PCSK9 revolution: Current status, controversies, and future directions. Trends Cardiovasc. Med. 2020, 30, 179–185.
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.G.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519.
- Yeang, C.; Cotter, B.; Tsimikas, S. Experimental Animal Models Evaluating the Causal Role of Lipoprotein(a) in Atherosclerosis and Aortic Stenosis. Cardiovasc. Drugs Ther. 2016, 30, 75–85.
- Chemello, K.; Beeské, S.; Tran, T.T.; Blanchard, V.; Villard, E.F.; Poirier, B.; Le Bail, J.-C.; Dargazanli, G.; Ho-Van-Guimbal, S.; Boulay, D.; et al. Lipoprotein(a) Cellular Uptake Ex Vivo and Hepatic Capture In Vivo Is Insensitive to PCSK9 Inhibition with Alirocumab. JACC Basic Transl. Sci. 2020, 5, 549–557, PMCID:PMC7315184.