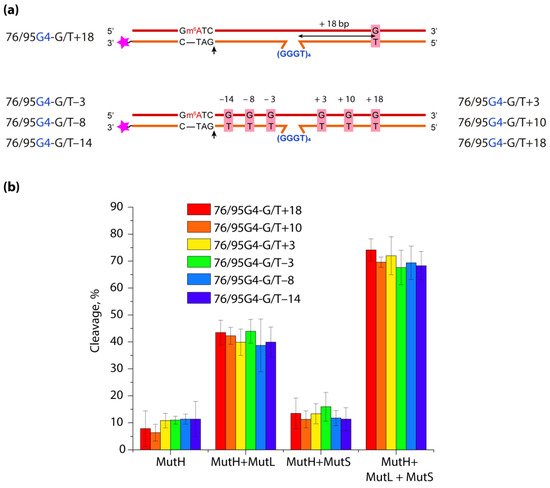
DNA G-quadruplexes (G4s) are known to be an integral part of the complex regulatory systems in both normal and pathological cells. Here we studied the effect of G4s on the DNA mismatch repair pathway. To assess the role of the distance between G4 and DNA mismatch on the functioning of the key mismatch repair protein, MutS, from E. coli on G4-containing substrates, a set of DNA duplexes with an embedded intramolecular parallel G4 structure and a monomethylated recognition site for the MutH endonuclease was prepared; the distance between the mismatched G/T pair and the G4 structure varied from 18 to 3 bp. It has been shown that this non-B form structure does not prevent activation of DNA mismatch repair even when the G4 structure and G/T pair are 3 bp apart. Thus, the preferential binding of MutS to G4 does not correlate with DNA mismatch repair activity, suggesting an unexpected role of these DNA-protein interactions in genome maintenance.
- DNA repair
- G-quadruplex-binding proteins
- DNA damage
- genomic instability
- MMR
- MutS
- MutL
- MutH
- Mismatch repair
1. Introduction
2. Influence of G-Quadruplexes on DNA Mismatch Repair
3. Conclusion
This entry is adapted from the peer-reviewed paper 10.3390/biom11091284
References
- Marušič, M.; Šket, P.; Bauer, L.; Viglasky, V.; Plavec, J. Solution-state structure of an intramolecular G-quadruplex with propeller, diagonal and edgewise loops. Nucleic Acids Res. 2012, 40, 6946–6956.
- Dolinnaya, N.G.; Ogloblina, A.M.; Yakubovskaya, M.G. Structure, properties, and biological relevance of the DNA and RNA G-quadruplexes: Overview 50 years after their discovery. Biochemistry 2016, 81, 1602–1649.
- Fujii, T.; Podbevšek, P.; Plavec, J.; Sugimoto, N. Effects of metal ions and cosolutes on G-quadruplex topology. J. Inorg. Biochem. 2017, 166, 190–198.
- Miyoshi, D. Structural transition from antiparallel to parallel G-quadruplex of d(G4T4G4) induced by Ca2+. Nucleic Acids Res. 2003, 31, 1156–1163.
- Chung, W.J.; Heddi, B.; Schmitt, E.; Lim, K.W.; Mechulam, Y.; Phan, A.T. Structure of a left-handed DNA G-quadruplex. Proc. Natl. Acad. Sci. USA 2015, 112, 2729–2733.
- Varizhuk, A.; Ischenko, D.; Tsvetkov, V.; Novikov, R.; Kulemin, N.; Kaluzhny, D.; Vlasenok, M.; Naumov, V.; Smirnov, I.; Pozmogova, G. The expanding repertoire of G4 DNA structures. Biochimie 2017, 135, 54–62.
- Guédin, A.; Gros, J.; Alberti, P.; Mergny, J.-L. How long is too long? Effects of loop size on G-quadruplex stability. Nucleic Acids Res. 2010, 38, 7858–7868.
- Mukundan, V.T.; Phan, A.T. Bulges in G-quadruplexes: Broadening the definition of G-quadruplex-forming sequences. J. Am. Chem. Soc. 2013, 135, 5017–5028.
- Chan, C.-Y.; Umar, M.I.; Kwok, C.K. Spectroscopic analysis reveals the effect of a single nucleotide bulge on G-quadruplex structures. Chem. Commun. 2019, 55, 2616–2619.
- Bao, H.-L.; Liu, H.; Xu, Y. Hybrid-type and two-tetrad antiparallel telomere DNA G-quadruplex structures in living human cells. Nucleic Acids Res. 2019, 47, 4940–4947.
- Kotar, A.; Rigo, R.; Sissi, C.; Plavec, J. Two-quartet kit* G-quadruplex is formed via double-stranded pre-folded structure. Nucleic Acids Res. 2019, 47, 2641–2653.
- Heddi, B.; Martín-Pintado, N.; Serimbetov, Z.; Kari, T.M.A.; Phan, A.T. G-quadruplexes with (4n-1) guanines in the G-tetrad core: Formation of a G-triad·water complex and implication for small-molecule binding. Nucleic Acids Res. 2016, 44, 910–916.
- Liu, H.; Wang, R.; Yu, X.; Shen, F.; Lan, W.; Haruehanroengra, P.; Yao, Q.; Zhang, J.; Chen, Y.; Li, S.; et al. High-resolution DNA quadruplex structure containing all the A-, G-, C-, T-tetrads. Nucleic Acids Res. 2018, 46, 11627–11638.
- Zhang, N.; Gorin, A.; Majumdar, A.; Kettani, A.; Chernichenko, N.; Skripkin, E.; Patel, D.J. Dimeric DNA quadruplex containing major groove-aligned A·T·A·T and G·C·G·C tetrads stabilized by inter-subunit Watson-Crick A·T and G·C pairs. J. Mol. Biol. 2001, 312, 1073–1088.
- Kolesnikova, S.; Hubálek, M.; Bednárová, L.; Cvačka, J.; Curtis, E.A. Multimerization rules for G-quadruplexes. Nucleic Acids Res. 2017, 45, 8684–8696.
- Bedrat, A.; Lacroix, L.; Mergny, J.-L. Re-evaluation of G-quadruplex propensity with G4Hunter. Nucleic Acids Res. 2016, 44, 1746–1759.
- Hänsel-Hertsch, R.; Beraldi, D.; Lensing, S.V.; Marsico, G.; Zyner, K.; Parry, A.; Di Antonio, M.; Pike, J.; Kimura, H.; Narita, M.; et al. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016, 48, 1267–1272.
- Todd, A.K.; Neidle, S. Mapping the sequences of potential guanine quadruplex motifs. Nucleic Acids Res. 2011, 39, 4917–4927.
- Huppert, J.L.; Balasubramanian, S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007, 35, 406–413.
- Dong, D.W.; Pereira, F.; Barrett, S.P.; Kolesar, J.E.; Cao, K.; Damas, J.; Yatsunyk, L.A.; Johnson, F.; Kaufman, B.A. Association of G-quadruplex forming sequences with human mtDNA deletion breakpoints. BMC Genom. 2014, 15, 677.
- Bartas, M.; Čutová, M.; Brázda, V.; Kaura, P.; Šťastný, J.; Kolomazník, J.; Coufal, J.; Goswami, P.; Červeň, J.; Pečinka, P. The presence and localization of G-quadruplex forming sequences in the domain of bacteria. Molecules 2019, 24, 1711.
- Zybailov, B.L.; Sherpa, M.D.; Glazko, G.V.; Raney, K.D.; Glazko, V.I. G4-quadruplexes and genome instability. Mol. Biol. 2013, 47, 197–204.
- Paeschke, K.; Bochman, M.L.; Garcia, P.D.; Cejka, P.; Friedman, K.L.; Kowalczykowski, S.C.; Zakian, V.A. Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature 2013, 497, 458–462.
- Biffi, G.; Tannahill, D.; McCafferty, J.; Balasubramanian, S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013, 5, 182–186.
- Laguerre, A.; Hukezalie, K.; Winckler, P.; Katranji, F.; Chanteloup, G.; Pirrotta, M.; Perrier-Cornet, J.-M.; Wong, J.M.Y.; Monchaud, D. Visualization of RNA-quadruplexes in live cells. J. Am. Chem. Soc. 2015, 137, 8521–8525.
- Henderson, A.; Wu, Y.; Huang, Y.C.; Chavez, E.A.; Platt, J.; Johnson, F.B.; Brosh, R.M.; Sen, D.; Lansdorp, P.M. Detection of G-quadruplex DNA in mammalian cells. Nucleic Acids Res. 2014, 42, 860–869.
- Shivalingam, A.; Izquierdo, M.A.; Le Marois, A.; Vyšniauskas, A.; Suhling, K.; Kuimova, M.K.; Vilar, R. The interactions between a small molecule and G-quadruplexes are visualized by fluorescence lifetime imaging microscopy. Nat. Commun. 2015, 6, 8178.
- Kotar, A.; Wang, B.; Shivalingam, A.; Gonzalez-Garcia, J.; Vilar, R.; Plavec, J. NMR Structure of a triangulenium-based long-lived fluorescence probe bound to a G-quadruplex. Angew. Chem. Int. Ed. 2016, 55, 12508–12511.
- Salgado, G.F.; Cazenave, C.; Kerkour, A.; Mergny, J.-L. G-quadruplex DNA and ligand interaction in living cells using NMR spectroscopy. Chem. Sci. 2015, 6, 3314–3320.
- Zhang, S.; Sun, H.; Wang, L.; Liu, Y.; Chen, H.; Li, Q.; Guan, A.; Liu, M.; Tang, Y. Real-time monitoring of DNA G-quadruplexes in living cells with a small-molecule fluorescent probe. Nucleic Acids Res. 2018, 46, 7522–7532.
- David, A.P.; Margarit, E.; Domizi, P.; Banchio, C.; Armas, P.; Calcaterra, N.B. G-quadruplexes as novel cis-elements controlling transcription during embryonic development. Nucleic Acids Res. 2016, 44, 4163–4173.
- Maizels, N.; Gray, L.T. The G4 genome. PLoS Genet. 2013, 9, e1003468.
- Pavlova, A.V., Monakhova, M.V., Ogloblina, A.M., Andreeva, N.A., Laptev, G.Y., Polshakov, V.I., Gromova, E.S., Zvereva, M.I., Yakubovskaya, M.G., Oretskaya, T.S., Kubareva, E.A., Dolinnaya, N.G. Responses of DNA mismatch repair proteins to a stable G-quadruplex embedded into a DNA duplex structure. Int. J. Mol. Sci. 2020, 21, 8773.
- Pavlova, A.V., Kubareva, E.A., Monakhova, M.V., Zvereva, M.I.; Dolinnaya, N.G.; Impact of G-Quadruplexes on the regulation of genome integrity, DNA damage and repair. Biomolecules 2021, 11, 1284, 10.3390/biom11091284.
- Lerner, L.K.; Sale, J.E. Replication of G quadruplex DNA. Genes 2019, 10, 95.
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637.
- Halabi, A.; Ditch, S.; Wang, J.; Grabczyk, E. DNA mismatch repair complex MutSβ promotes GAA·TTC repeat expansion in human cells. J. Biol. Chem. 2012, 287, 29958–29967.
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346.
- Perevoztchikova, S.A.; Romanova, E.A.; Oretskaya, T.S.; Friedhoff, P.; Kubareva, E.A. Modern aspects of the structural and functional organization of the DNA mismatch repair system. Acta Nat. 2013, 5, 17–34.
- Ehrat, E.A.; Johnson, B.R.; Williams, J.D.; Borchert, G.M.; Larson, E.D. G-quadruplex recognition activities of E. coli MutS. BMC Mol. Biol. 2012, 13, 23.
- Larson, E.D.; Duquette, M.L.; Cummings, W.J.; Streiff, R.J.; Maizels, N. MutSα binds to and promotes synapsis of transcriptionally activated immunoglobulin switch regions. Curr. Biol. 2005, 15, 470–474.
- Liu, J.; Hanne, J.; Britton, B.M.; Bennett, J.; Kim, D.; Lee, J.-B.; Fishel, R. Cascading MutS and MutL sliding clamps control DNA diffusion to activate mismatch repair. Nature 2016, 539, 583–587.
- Evans, E.; Alani, E. Roles for mismatch repair factors in regulating genetic recombination. Mol. Cell. Biol. 2000, 20, 7839–7844.
- Spies, M.; Fishel, R. Mismatch repair during homologous and homeologous recombination. Cold Spring Harb. Perspect. Biol. 2015, 7, a022657.
- Fishel, R. Mismatch repair. J. Biol. Chem. 2015, 290, 26395–26403.