Air pollutants include toxic particles and gases emitted in large quantities from many different combustible materials. They also include particulate matter (PM) and ozone, and biological contaminants, such as viruses and bacteria, which can penetrate the human airway and reach the bloodstream, triggering airway inflammation, dysfunction, and fibrosis.
1. Introduction
Air pollutants include toxic particles and gases emitted in large quantities from many different sources, including vehicles and factories [
1]. Indoor pollutants include smoke from tobacco, cooking, and the burning of wood and other materials in stoves and fireplaces, as well as dust particles disturbed during cleaning and outdoor particles that infiltrate the indoor environment [
1]. Major pollutants include particulate matter (PM), ozone, nitrogen dioxide (NO
2), and sulfur dioxide (SO
2). Biological contaminants such as viruses, bacteria, animal dander and cat saliva, house dust mites (HDMs), cockroaches, and pollen can exacerbate allergic reactions and airway diseases, such as asthma, allergic rhinitis (AR), and hypersensitivity pneumonitis [
2,
3,
4].
2. The Effects of Air Pollutants on Asthma and COPD, Upper Airway Disease
Air pollution is one of the most important environmental factors affecting public health, due to its effects on the respiratory system [40]. Air pollution can interfere with defense mechanisms in the lung, weaken the body’s immune response [41], and trigger oxidative stress and inflammation [42,43]. Air pollution is defined as the presence of aerial substances that are harmful to humans, and is associated with a higher risk of premature death due to cardiovascular diseases (e.g., ischemic heart disease and stroke), asthma, COPD, lower respiratory tract infections, and lung cancer [31,40]. Ozone has a strong smell and irritates the respiratory system, resulting in swelling of the throat, discomfort in the chest, coughing, sputum production, and even emphysema with long-term exposure [44]. Nitric oxide may generate photochemical smog and has acute toxic effects on the human lungs. The short- and long-term effects of PM10, PM2.5, and SO2 on lung function, disease morbidity, and mortality have been described in detail [23,40,45]. Air pollution is associated with various respiratory and non-respiratory diseases including asthma, COPD, pneumonia, lung malignancies, heart disease, stroke, dementia, and diabetes [46,47,48,49,50].
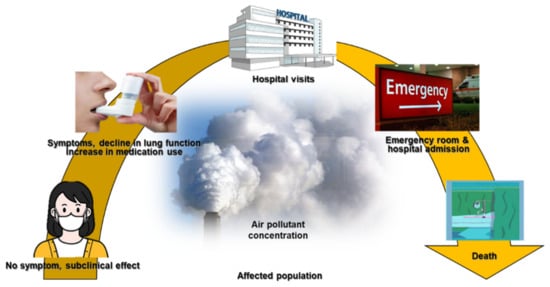
The long-term effects of air pollution on asthma were summarized in an American Thoracic Society workshop report, which indicated that long-term exposure to air pollution was a cause of childhood asthma. However, there was insufficient evidence to draw a similar conclusion regarding adult asthma [
65]. Many studies have described correlations between short-term exposure to outdoor air pollutants and various aspects of asthma, including symptom control [
66], lung function [
67], medication dose [
68,
69], outpatient visits [
70,
71], asthma exacerbations [
72,
73], emergency room visits [
74], hospitalizations [
75,
76], length of hospital stay [
77], and mortality rate [
78] (
Figure 1).
Figure 1. The impact of air pollutants on respiratory diseases.
The most common cause of COPD is smoking; some cases are also due to air pollution and genetics [82]. Poorly ventilated cooking fires, often using coal or biomass fuels such as wood, lead to indoor air pollution and are a common cause of COPD in developing countries [83]. Such fires are used by nearly 3 billion people for cooking and heating, and adverse effects on health are more frequent in women due to their higher levels of exposure [84,85]. These fires are the main source of energy in 80% of all homes in India, China, and sub-Saharan Africa [85].
Several epidemiological studies have shown that air pollutants exacerbate airway diseases such as AR, asthma, bronchitis, and COPD. Pollutants such as TRAPs also have negative effects on other upper airway diseases such as AR and non-AR, sinusitis, and otitis media [87]. Increasing evidence suggests that PM, photochemical pollutants, and ozone are also linked to the development of upper airway diseases [87]. Young children and individuals who are obese are particularly susceptible to these conditions [87]. ROS, apoptosis, and inflammation are all involved in the pathophysiological etiology of upper airway diseases [87]. Although the data conflict, and controlled prospective studies are needed to determine the relevant mechanisms and risk factors, traffic fumes and tobacco smoke are major factors exacerbating upper airway diseases [87].
3. Airway Toxicity Mechanisms Related to Air Pollutants
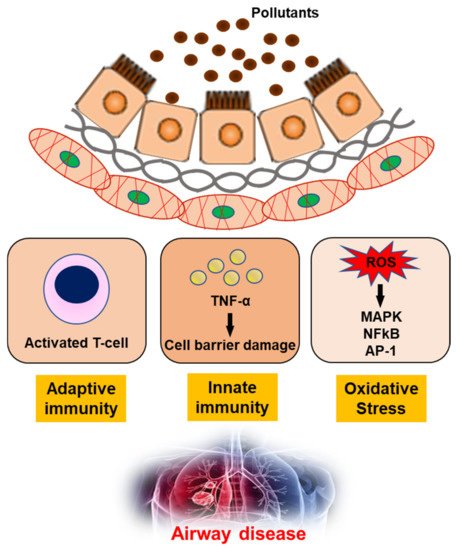
The impact of air pollution, especially on population health, cannot be underestimated and urgently needs to be addressed [
93]. In-depth studies of the underlying mechanisms, together with clinical, imaging and molecular biology data, have shown that exposure to air pollution promotes the development of airway diseases [
94,
95,
96,
97]. Pollutants can generate oxidant-mediated cellular damage [
28] via ROS production, other types of oxidative stress, and innate and adaptive immune responses that may have adverse effects on health (
Figure 2).
Figure 2. The mechanism of air pollutants for airway disease. TNF-α; Tumor necrosis factor-α, ROS; reactive oxygen species, MAPK; mitogen-activated protein kinase, NFkB; nuclear factor kappa-light-chain-enhancer of activated B cells, AP-1; Activator protein 1.
Ozone is highly reactive, and oxidizes proteins and lipids in the fluid-lined compartment of the lung. This initiates inflammation and increases lung permeability, via cytotoxic mediators including pro-inflammatory cytokines, ROS, and nitrogen intermediates such as peroxynitrite [
17]. The primary targets for ozone are the distal structures of the lung, including the terminal bronchioles, bronchiole–alveolar duct junction, and proximal alveolar regions [
17]. Acute inhalation of ozone causes structural alterations in the lung, including disruption of the alveolar epithelial barrier, which lead to alveolar epithelial type II cell hypertrophy and hyperplasia [
93,
98]. The recruitment of inflammatory cells into the lung following ozone exposure can also damage tissue via the release of toxic mediators (e.g., cytokines, ROS, nitrogen species, and proteolytic enzymes) from activated macrophages and neutrophils [
93,
98].
Little is known about the combined effect of ozone and diesel exhaust particles (DEPs) on the development and exacerbation of asthma. Co-exposure to ozone and DEPs has an additive effect on AHR, exerted via the modulation of interleukin (IL)-4 and interferon (IFN)-γ, suggesting that DEPs amplify the Th2 immune response [
102]. The AHR induced by acute inhalation of ozone depends on the concentration of ozone and duration of exposure. Airway obstruction is induced following ozone exposure in a concentration-dependent manner and persists for at least 72 h [
103,
104].
PM is the main component of most air pollutants. PM includes a range of particle sizes, such as coarse, fine, and ultrafine particles. Individuals are primarily exposed to PM via inhalation. The inhalation of PM exacerbates respiratory symptoms in patients with chronic airway diseases, but the mechanisms underlying this response remain unclear [
16]. Nanoparticles (NP) may cause cell and tissue damage, leading to local and systemic inflammatory responses and adverse effects on health. The inflammasome is a major regulator of inflammation via its activation of pro-caspase-1, which cleaves pro-IL-1β into a mature form and may induce acute and chronic immune responses to NPs. Inflammasome activation was observed in the lungs of individuals with asthma following NP exposure, suggesting that targeting the inflammasome may assist in controlling NP-induced airway inflammation and hyperresponsiveness [
106]. Interestingly, treatment with titanium dioxide (TiO
2) increased the level of mRNA-encoding macrophage migration-inhibitory factor (MIF). MIF was primarily expressed in the epithelium and was elevated in lung tissue and bronchoalveolar lavage fluid from TiO
2- compared to sham-treated rats. Carbon black and DEPs also induced the expression of MIF protein in epithelial cells [
107].
DEPs can trigger AHR and inflammation. Long-term DEP exposure increased AHR, inflammation, lung fibrosis, and goblet cell hyperplasia in a mouse model [
108]. The interaction between chronic inflammation and neural dysfunction in the airways suggests a link between the nervous and immune systems. Substance P, ATP, and calcitonin gene-related peptide (CGRP) levels in bronchoalveolar lavage fluid were increased in ovalbumin (OVA)-sensitized mice, and these increases were augmented in OVA-sensitized NP-exposed mice. Bradykinin, ATP, and CGRP levels were all increased in NP-exposed normal human bronchial epithelial (NHBE) cells in a dose-dependent manner. Calcium concentrations were increased in NHBE cells exposed to NPs for 8 h. These results indicate that neuroinflammation may be involved in the pathogenesis of bronchial asthma, and that NPs can exacerbate asthma via neuro-mediator release [
109].
Unsurprisingly, exposure to environmental pollutants is associated with adverse respiratory outcomes. The phosphorylation of enzymes activates or deactivates many cellular processes, and is linked to the development of lung diseases such as asthma and COPD. Immunoblotting with anti-GSTP1 antibody revealed no change in GSTP1 protein levels in BEAS-2B cell lysates after treatment with TiO
2 particles; blotting with anti-phosphoserine and anti-phosphotyrosine antibodies revealed dose-dependent decreases in phosphoserine and phosphotyrosine proteins. Exposure to foreign particles phosphorylated and dephosphorylated several proteins within epithelial cells, and the serine and tyrosine phosphorylation levels of GSTP1 decreased. These data indicate that airborne particles affect the pattern of phosphorylation of proteins involved in defense and apoptosis within the respiratory epithelium [
111].
Chitinase may play a regulatory role in allergic diseases. In a mouse model, DEPs induced AHR and Ym1/2 mRNA expression via a Th2-cell-mediated response, suggesting that chitinase may play an important role in airway inflammation and responsiveness upon exposure to DEPs, and may, therefore, be involved in regulating allergic diseases [
112].
Several allergic and inflammatory diseases are reportedly associated with epithelial barrier defects and TJ disruption, including atopic dermatitis, asthma, and chronic rhinosinusitis. TJs may also play a role in asthma development [
117]. However, little is known of the interplay between air pollutants and the bronchial epithelial barrier, or of the impact of PM on epithelial barrier function. PM-induced disruption of barrier function in a bronchial epithelial cell line has provided some insight into the role of epithelial barrier dysfunction in asthma [
118]. When PM is inhaled, NPs such as TiO
2 particles may cause cell and tissue damage, leading to local and systemic inflammatory responses and adverse effects on health [
119]. The bronchial epithelium is constantly exposed to a wide range of environmental substances present in inhaled air, including noxious gases and anthropogenic and natural particles. These include gases and particles from car emissions, tobacco smoke, pollen, animal dander, and pathogens [
120]. Therefore, the airway epithelium is an important physical barrier, as well as a modulator of allergic responses and airway inflammation [
106]. Epithelial barrier dysfunction contributes to allergic inflammation and the development of asthma, because a dysfunctional barrier increases the exposure of subepithelial tissues to inhaled allergens and air pollutants. The TJ proteins known as claudins (CLDNs) are important regulators of paracellular permeability. PM exacerbates airway epithelial barrier dysfunction and leads to airway inflammation [
121].
The airway epithelium is the initial barrier to external pathogens, including bacteria, viruses, chemical substances, and allergens [
122,
123,
124]. Airway epithelial cells also have pivotal roles as coordinators of immunological defense and mediate the elimination of external pathogens from airways. When the airway epithelium is damaged, pathogens can remain in the airway and induce aberrant immunological reactions [
122,
123,
124]. Dysregulated function of the asthmatic airway epithelium reportedly interferes with wound repair, weakens TJs, and leads to excessive proliferation and airway remodeling. This results in aberrant airway responses to external pathogens [
122,
123,
124].
Functional studies indicate that asthmatic airway epithelia show increased sensitivity to environmental stressors and oxidative stress, thus reducing the threshold for epithelial damage [
125,
126,
127]. Increased barrier permeability in patients with asthma increases susceptibility to allergens, reduces the threshold for epithelial damage, and activates type 2 responses [
128,
129,
130,
131]. Changes in microbial diversity within asthmatic airways have also been reported [
128,
129,
130,
131]. Furthermore, impaired epithelial barrier repair in patients with asthma can weaken inflammatory responses [
132,
133].
Levels of the TJ protein CLDN7 were decreased in the plasma of patients with asthma. In these patients, CLDN7 levels were indicators of lung function and the blood eosinophil concentration. CLDN7 expression was elevated in the lungs of mice with asthma, and in NHBE cells treated with HDM extracts. However, CLDN7 expression was suppressed by exposure to TiO
2. P-AKT and p-ERK were increased in asthmatic mice and decreased in those treated with TiO
2. Levels of p-AKT and p-ERK were decreased in NHBE cells treated with TiO
2 and HDM extracts. Furthermore, transepithelial electrical resistance increased in NHBE cells treated with TiO
2 or HDM extracts. However, this effect was attenuated when TiO
2 and HDM extracts were co-administered. These data suggest that PM exacerbates airway epithelial barrier dysfunction and leads to airway inflammation [
121].
The expression patterns of CLDN-4, -5, and -17 were different in the nose and lungs of mice exposed to DEPs. This suggests that similar changes occur in the cell barriers lining the upper and lower airways, raising the possibility that modulating cell barriers in the nose and lung may be useful for treating diseases of the airway [
134].
N-acetylcysteine (NAC) affects the signaling pathways that are involved in apoptosis, angiogenesis, cell growth and arrest, redox-regulated gene expression, and the inflammatory response. NAC attenuated OVA-induced AHR and inflammation. Levels of CLDN18 protein in lung tissue from OVA-sensitized mice were higher than those in lung tissue from control mice, and they increased in response to treatment with NAC or dexamethasone. Treatment with NAC or dexamethasone also suppressed the OVA-induced increase in IL-1α protein levels. Although treatment with NAC increased OVA-induced p-PDK1 protein levels, it decreased phosphorylated Akt (pAkt)/Akt levels. Therefore, CLDN18 plays an important role in the pathogenesis of asthma and NAC diminishes airway inflammation by modulating CLDN18 expression [
135].
CLDNs are major transmembrane protein components of TJs in the endothelia and epithelia. They enable TJs to maintain cell permeability, and also facilitate cell signaling via protein–protein interactions. CLDNs are implicated in airway inflammation following ozone exposure, suggesting that ozone affects TJ proteins via oxidative stress [
136].
Inhaled corticosteroids are the most effective anti-inflammatory therapy currently available to treat persistent asthma. Corticosteroid therapy can also attenuate the increases in bronchial vascularity and edema frequently observed in patients with asthma. In our inflamed-airway mouse model, AHR and cytokine levels were reduced by corticosteroid treatment, and abnormal CLDN5 expression and endothelial integration were attenuated; this suggests that endothelial TJs may be therapeutic targets for decreasing airway inflammation. These findings indicate that the regulation of lung endothelial barrier function may be a promising novel therapeutic approach to treating asthma [
138].
CLDN5 is critical for controlling endothelial cellular polarity and pericellular permeability. Mean plasma CLDN5 levels were higher in patients with COPD exacerbations than in those with stable COPD. The plasma CLDN5 levels measured in patients with COPD correlated with the duration of smoking. Plasma CLDN5 levels in patients with stable COPD were correlated with the predicted FEV1%d Plasma CLDN5 levels were not correlated with age. CLDN5 may be involved in COPD pathogenesis [
139].
Respiratory syncytial virus (RSV) is the leading cause of lower respiratory tract infections in children worldwide. Although most of those affected develop a mild self-limiting illness, some develop severe acute lower respiratory infections and persistent airway disease. Exposure to ambient PM has been linked to asthma, bronchitis, and viral infections in many epidemiological studies. TiO
2-NP exposure exacerbates RSV-induced adherens junction (AJ) complex dysfunction, and also exacerbates inflammation by generating ROS [
140].
Airway epithelial barrier function is maintained by the formation of TJs and AJs. Inhalation of cigarette smoke (CS) causes airway epithelial barrier dysfunction and may exacerbate the pathogenesis of chronic lung diseases, such as asthma and COPD. Treatment with CS extract resulted in airway epithelial barrier dysfunction, and also downregulated many TJ and AJ proteins. LL-37 counteracted CS extract-induced reductions in transepithelial resistance and prevented the disruption of occludin and ZO-1. Therefore, using LL-37 to counteract airway epithelial barrier dysfunction may benefit patients with respiratory diseases such as asthma and COPD [
141].
CLDN5 is a critical component of the endothelial TJs, which control pericellular permeability. Acrolein, as one of the major irritants present in smoke, can induce acute lung injury, possibly by altering CLDN5 expression. Lung CLDN5 transcript and protein levels increased more in an acrolein-resistant cell line than in a susceptible cell line. In human endothelial cells, 30 nM acrolein increased CLDN5 transcript and p-FOXO1 protein levels. The phosphatidylinositol 3-kinase inhibitor LY294002 attenuated acrolein-induced increases in CLDN5 transcript levels. Therefore, maintaining endothelial CLDN5 levels may be a novel clinical approach to treating acute lung injury [
144].
Acrolein, an α/β-unsaturated aldehyde, is volatile at room temperature. It is a respiratory irritant present in environmental tobacco smoke and may be generated during cooking or at endogenous injury sites. Acrolein induces reactive airway dysfunction syndrome (RADS). Mouse model studies demonstrated that ROS, angiogenesis, and TJ proteins were involved in the development of RADS [
145,
146].
COPD pathogenesis is driven by the airway epithelium [
147]. One important factor is a disease-related reduction in barrier function, which is potentiated by the dysregulation of TJ protein complexes [
147]. The epithelial airway lining forms the initial barrier to environmental particles, such as inhaled CS, which is a major risk factor in COPD development [
147]. The barrier is formed by epithelial junctions, which are interconnected structures that restrict the access of inhaled pathogens and environmental stressors [
147].
Taken together, the data showed that ApoA1 overexpression attenuated CS-induced lung inflammation and emphysema in mice. Therefore, targeted augmentation of ApoA1 in the lung may have therapeutic potential by preventing smoking-related COPD/emphysema [
148].
Acute exacerbations of COPD may occur due to air pollution, and ozone is an important pollutant. Vimentin, lactate dehydrogenase A, and triose phosphate isomerase levels were all decreased by both smoking and ozone exposure treatment.
4. Conclusions
Air pollution such as ozone, NO2, SO2, and PM is a major public health problem that exacerbates airway diseases such as asthma, COPD, and AR. Airway epithelial cells form the initial barrier to pollutants and play pivotal roles in the development of asthma and COPD. These cells are also potential therapeutic targets for maintaining airway integrity. Further basic and clinical research is warranted to identify environmental pollutants and novel therapeutic targets, as well as to elucidate their underlying mechanisms.
This entry is adapted from the peer-reviewed paper 10.3390/ijerph18189905