Autoimmune polyendocrine syndrome type 1 (APS-1) is a rare but severe monogenetic autoimmune endocrine disease caused by failure of the Autoimmune Regulator (AIRE). AIRE regulates the negative selection of T cells in the thymus, and the main pathogenic mechanisms are believed to be T cell-mediated, but little is known about the role of B cells.
- autoimmune polyendocrine syndrome type 1 (APS-1)
- autoantibodies
- B-cells
- B-cell dependent therapy
- mouse models of Aire deficiency
1. Introduction
Autoimmune polyendocrine syndrome type 1 (APS-1), also known as autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy (APECED), is a monogenetic autoimmune disease with an estimated prevalence of 1:100,000 caused by mutations in the autoimmune regulator ( AIRE ) gene [1][2][3][4][5]. Patients usually develop autoimmune manifestations in multiple organs leading to functional impairment, especially of various endocrine glands. APS-1 is diagnosed by the presence of two out of three major clinical components of hypoparathyroidism, chronic mucocutaneous candidiasis (CMC), and primary adrenocortical insufficiency or Addison’s disease. Besides these, multiple other manifestations occur, like primary ovarian insufficiency (POI), autoimmune thyroid disease, type 1 diabetes (T1D), autoimmune gastritis, keratitis, vitiligo, alopecia and enamel hypoplasia are common [1][6]. Alternatively, finding AIRE mutations and specific autoantibodies can be used diagnostically and enable early diagnosis before the main components develop [2][7].
The disease-causing mutations in AIRE are typically inherited in an autosomal recessive manner [8][9], although several heterozygous missense mutations have also been found with a dominant-negative inheritance pattern [10][11]. Overall, more than 130 mutations in AIRE have been identified (Human Gene Mutation Database, www.hgmd.cf.ac.uk , accessed on the 10 of May 2021), many of which cluster in key domains of the AIRE protein [2][8][12][13][14][15][16]. AIRE is mainly expressed in a subset of thymic medullary epithelial cells (mTECs), regulating the expression of 20% of the 20,000 unique tissue-restricted antigens (TRAs) to be presented to the developing T cells during negative selection [17][18][19][20][21]. This transcription factor contributes to the development of thymic Foxp3+ CD4+ regulatory T cells (Tregs) and is crucial for their ability to re-circulate back to the thymus [22][23][24]. In addition, AIRE is necessary for the generation of Tregs ex-thymus [25]. Indeed, APS-1 patients have decreased numbers of Tregs with a modified TCR repertoire compared to healthy individuals, reflecting the abnormal selection of T cells in the thymus [26][27].
Although the pathogenic mechanisms in APS-1 are T cell-mediated, the B cells are important antigen-presenting cells (APCs) that rely on T cell activation for the production of antibodies. They are found in the thymus and are also reported to have some AIRE expression themselves [28][29][30]. We will here look closer into what is known about the B cells in APS-1 patients and AIRE-deficient mouse models, summarize the status quo and the outstanding research questions, and highlight the therapeutic strategies involving B cells in APS-1.
B Cell’s Contribution to APS-1 and Aire Deficiency
2. Treatment Approaches Targeting B Cells in Autoimmune Diseases
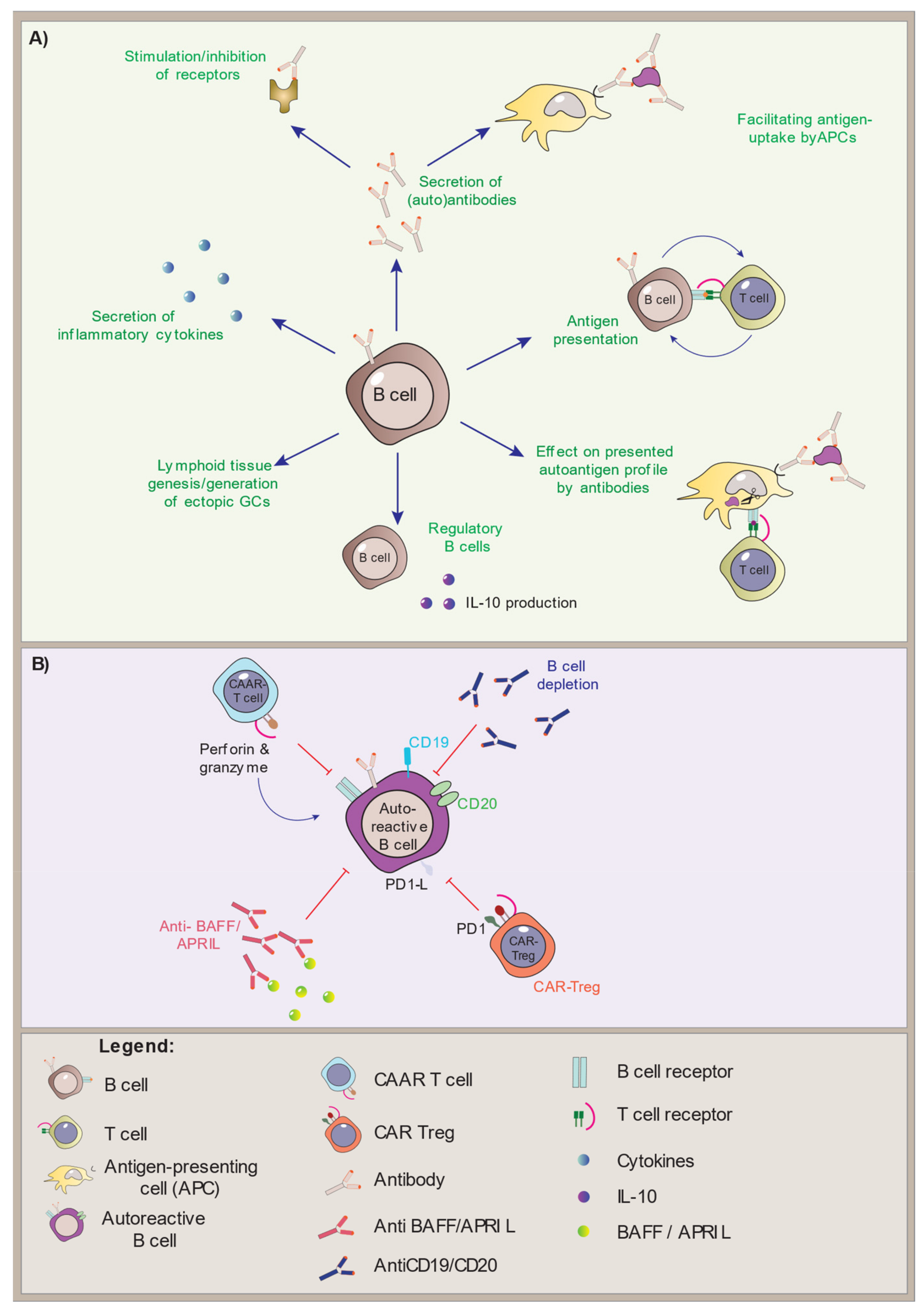
3. Conclusions and Future Perspectives
-
What is the inflammatory cytokine secretion profile of B cells in APS-1 patients?
-
How do B cells in APS-1 patients interact with T cells, DCs, and macrophages, especially with regards to antigen presentation?
-
Is the Breg subset functional in APS-1?
-
Are the hallmark autoantibodies in plasma and sera from APS-1 patients pathogenic?
-
Does B cell depletion therapy improve the main manifestations, and does it impact the interferon profile?
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines9091274
References
- Husebye, E.S.; Anderson, M.S.; Kämpe, O. Autoimmune Polyendocrine Syndromes. N. Engl. J. Med. 2018, 378, 1132–1141.
- Wolff, A.S.B.; Erichsen, M.M.; Meager, A.; Magitta, N.F.; Myhre, A.G.; Bollerslev, J.; Fougner, K.J.; Lima, K.; Knappskog, P.M.; Husebye, E.S. Autoimmune Polyendocrine Syndrome Type 1 in Norway: Phenotypic Variation, Autoantibodies, and Novel Mutations in the Autoimmune Regulator Gene. J. Clin. Endocrinol. Metab. 2007, 92, 595–603.
- Ahonen, P.; Myllärniemi, S.; Sipilä, I.; Perheentupa, J. Clinical Variation of Autoimmune Polyendocrinopathy–Candidiasis–Ectodermal Dystrophy (APECED) in a Series of 68 Patients. N. Engl. J. Med. 1990, 322, 1829–1836.
- Perheentupa, J. Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy. J. Clin. Endocrinol. Metab. 2006, 91, 2843–2850.
- Zlotogora, J.; Shapiro, M.S. Polyglandular autoimmune syndrome type I among Iranian Jews. J. Med. Genet. 1992, 29, 824–826.
- Guo, C.-J.; Leung, P.S.; Zhang, W.; Ma, X.; Gershwin, M.E. The immunobiology and clinical features of type 1 autoimmune polyglandular syndrome (APS-1). Autoimmun. Rev. 2018, 17, 78–85.
- Meloni, A.; Furcas, M.; Cetani, F.; Marcocci, C.; Falorni, A.; Perniola, R.; Pura, M.; Wolff, A.S.B.; Husebye, E.S.; Lilic, D.; et al. Autoantibodies against Type I Interferons as an Additional Diagnostic Criterion for Autoimmune Polyendocrine Syndrome Type I. J. Clin. Endocrinol. Metab. 2008, 93, 4389–4397.
- Aaltonen, J.; Björses, P.; Perheentupa, J.; Horelli–Kuitunen, N.; Palotie, A.; Peltonen, L.; Lee, Y.S.; Francis, F.; Henning, S.; Thiel, C.; et al. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat. Genet. 1997, 17, 399–403.
- Nagamine, K.; Peterson, P.; Scott, H.; Kudoh, J.; Minoshima, S.; Heino, M.; Krohn, K.J.E.; Lalioti, M.D.; Mullis, P.E.; Antonarakis, S.; et al. Positional cloning of the APECED gene. Nat. Genet. 1997, 17, 393–398.
- Oftedal, B.; Hellesen, A.; Erichsen, M.M.; Bratland, E.; Vardi, A.; Perheentupa, J.; Kemp, E.H.; Fiskerstrand, T.; Viken, M.K.; Weetman, A.P.; et al. Dominant Mutations in the Autoimmune Regulator AIRE Are Associated with Common Organ-Specific Autoimmune Diseases. Immunity 2015, 42, 1185–1196.
- Cetani, F.; Barbesino, G.; Borsari, S.; Pardi, E.; Cianferotti, L.; Pinchera, A.; Marcocci, C. A Novel Mutation of the Autoimmune Regulator Gene in an Italian Kindred with Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy, Acting in a Dominant Fashion and Strongly Cosegregating with Hypothyroid Autoimmune Thyroiditis. J. Clin. Endocrinol. Metab. 2001, 86, 4747–4752.
- Abramson, J.; Husebye, E.S. Autoimmune regulator and self-tolerance - molecular and clinical aspects. Immunol. Rev. 2016, 271, 127–140.
- Bruserud, Ø.; Oftedal, B.E.; Wolff, A.B.; Husebye, E.S. AIRE-mutations and autoimmune disease. Curr. Opin. Immunol. 2016, 43, 8–15.
- Björses, P.; Halonen, M.; Palvimo, J.; Kolmer, M.; Aaltonen, J.; Ellonen, P.; Perheentupa, J.; Ulmanen, I.; Peltonen, L. Mutations in the AIRE Gene: Effects on Subcellular Location and Transactivation Function of the Autoimmune Polyendocrinopathy-Candidiasis–Ectodermal Dystrophy Protein. Am. J. Hum. Genet. 2000, 66, 378–392.
- Stolarski, B.; Pronicka, E.; Korniszewski, L.; Pollak, A.; Kostrzewa, G.; Rowińska, E.; Włodarski, P.; Skórka, A.; Gremida, M.; Krajewski, P.; et al. Molecular background of polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome in a Polish population: Novel AIRE mutations and an estimate of disease prevalence. Clin. Genet. 2006, 70, 348–354.
- Pearce, S.H.; Cheetham, T.; Imrie, H.; Vaidya, B.; Barnes, N.D.; Bilous, R.W.; Carr, D.; Meeran, K.; Shaw, N.J.; Smith, C.S.; et al. A Common and Recurrent 13-bp Deletion in the Autoimmune Regulator Gene in British Kindreds with Autoimmune Polyendocrinopathy Type 1. Am. J. Hum. Genet. 1998, 63, 1675–1684.
- Goodnow, C.; Sprent, J.; Groth, B.F.D.S.; Vinuesa, C. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nat. Cell Biol. 2005, 435, 590–597.
- Anderson, M.S.; Venanzi, E.; Chen, Z.; Berzins, S.P.; Benoist, C.; Mathis, D. The Cellular Mechanism of Aire Control of T Cell Tolerance. Immunity 2005, 23, 227–239.
- Takahama, Y. Journey through the thymus: Stromal guides for T-cell development and selection. Nat. Rev. Immunol. 2006, 6, 127–135.
- Anderson, M.S.; Venanzi, E.S.; Klein, L.; Chen, Z.; Berzins, S.P.; Turley, S.J.; von Boehmer, H.; Bronson, R.; Dierich, A.; Benoist, C.; et al. Projection of an Immunological Self Shadow Within the Thymus by the Aire Protein. Science 2002, 298, 1395–1401.
- Sansom, S.N.; Shikama-Dorn, N.; Zhanybekova, S.; Nusspaumer, G.; Macaulay, I.; Deadman, M.E.; Heger, A.; Ponting, C.; Holländer, G.A. Population and single-cell genomics reveal theAiredependency, relief from Polycomb silencing, and distribution of self-antigen expression in thymic epithelia. Genome Res. 2014, 24, 1918–1931.
- Cowan, J.E.; Baik, S.; McCarthy, N.I.; Parnell, S.M.; White, A.J.; Jenkinson, W.E.; Anderson, G. Aire controls the recirculation of murine Foxp3+regulatory T-cells back to the thymus. Eur. J. Immunol. 2018, 48, 844–854.
- Malchow, S.; Leventhal, D.S.; Lee, V.; Nishi, S.; Socci, N.D.; Savage, P.A. Aire Enforces Immune Tolerance by Directing Autoreactive T Cells into the Regulatory T Cell Lineage. Immunity 2016, 44, 1102–1113.
- Yang, S.; Fujikado, N.; Kolodin, D.; Benoist, C.; Mathis, D. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science 2015, 348, 589–594.
- Zhang, X.; Li, X.C.; Xiao, X.; Sun, R.; Tian, Z.; Wei, H. CD4+CD62L+ Central Memory T Cells Can Be Converted to Foxp3+ T Cells. PLoS ONE 2013, 8, e77322.
- Sng, J.; Ayoglu, B.; Chen, J.W.; Schickel, J.-N.; Ferre, E.M.N.; Glauzy, S.; Romberg, N.; Hoenig, M.; Cunningham-Rundles, C.; Utz, P.J.; et al. AIRE expression controls the peripheral selection of autoreactive B cells. Sci. Immunol. 2019, 4, eaav6778.
- Koivula, T.-T.; Laakso, S.M.; Niemi, H.J.; Kekäläinen, E.; Laine, P.; Paulin, L.; Auvinen, P.; Arstila, T.P. Clonal Analysis of Regulatory T Cell Defect in Patients with Autoimmune Polyendocrine Syndrome Type 1 Suggests Intrathymic Impairment. Scand. J. Immunol. 2017, 86, 221–228.
- Gies, V.; Guffroy, A.; Danion, F.; Billaud, P.; Keime, C.; Fauny, J.-D.; Susini, S.; Soley, A.; Martin, T.; Pasquali, J.-L.; et al. B cells differentiate in human thymus and express AIRE. J. Allergy Clin. Immunol. 2017, 139, 1049–1052.e12.
- Yamano, T.; Nedjic, J.; Hinterberger, M.; Steinert, M.; Koser, S.; Pinto, S.; Gerdes, N.; Lutgens, E.; Ishimaru, N.; Busslinger, M.; et al. Thymic B Cells Are Licensed to Present Self Antigens for Central T Cell Tolerance Induction. Immunity 2015, 42, 1048–1061.
- Suzuki, E.; Kobayashi, Y.; Kawano, O.; Endo, K.; Haneda, H.; Yukiue, H.; Sasaki, H.; Yano, M.; Maeda, M.; Fujii, Y. Expression of AIRE in thymocytes and peripheral lymphocytes. Autoimmunity 2008, 41, 133–139.
- Perri, V.; Gianchecchi, E.; Scarpa, R.; Valenzise, M.; Rosado, M.M.; Giorda, E.; Crinò, A.; Cappa, M.; Barollo, S.; Garelli, S.; et al. Altered B cell homeostasis and Toll-like receptor 9-driven response in patients affected by autoimmune polyglandular syndrome Type 1. Immunobiology 2017, 222, 372–383.
- Zhao, B.; Chang, L.; Fu, H.; Sun, G.; Yang, W. The Role of Autoimmune Regulator (AIRE) in Peripheral Tolerance. J. Immunol. Res. 2018, 2018.
- Zhu, W.; Yang, W.; He, Z.; Liao, X.; Wu, J.; Sun, J.; Yang, Y.; Li, Y. Overexpressing autoimmune regulator regulates the expression of toll-like receptors by interacting with their promoters in RAW264.7 cells. Cell. Immunol. 2011, 270, 156–163.
- Gavanescu, I.; Benoist, C.; Mathis, D. B cells are required for Aire-deficient mice to develop multi-organ autoinflammation: A therapeutic approach for APECED patients. Proc. Natl. Acad. Sci. USA 2008, 105, 13009–13014.
- Popler, J.; Alimohammadi, M.; Kämpe, O.; Dalin, F.; Dishop, M.K.; Barker, J.M.; Moriarty-Kelsey, M.; Soep, J.B.; Deterding, R.R. Autoimmune polyendocrine syndrome type 1: Utility of KCNRG autoantibodies as a marker of active pulmonary disease and successful treatment with rituximab. Pediatr. Pulmonol. 2011, 47, 84–87.
- Kato, A.; Hulse, K.; Tan, B.K.; Schleimer, R.P. B-lymphocyte lineage cells and the respiratory system. J. Allergy Clin. Immunol. 2013, 131, 933–957.
- Napier, C.; Gan, E.H.; Mitchell, A.L.; Gilligan, L.C.; Rees, D.A.; Moran, C.; Chatterjee, K.; Vaidya, B.; James, R.A.; Mamoojee, Y.; et al. Residual Adrenal Function in Autoimmune Addison’s Disease—Effect of Dual Therapy With Rituximab and Depot Tetracosactide. J. Clin. Endocrinol. Metab. 2019, 105, e1250–e1259.
- Pearce, S.H.S.; Mitchell, A.L.; Bennett, S.; King, P.; Chandran, S.; Nag, S.; Chen, S.; Smith, B.R.; Isaacs, J.; Vaidya, B. Adrenal Steroidogenesis after B Lymphocyte Depletion Therapy in New-Onset Addison’s Disease. J. Clin. Endocrinol. Metab. 2012, 97, E1927–E1932.
- Devoss, J.J.; Shum, A.K.; Johannes, K.P.A.; Lu, W.; Krawisz, A.K.; Wang, P.; Yang, T.; LeClair, N.P.; Austin, C.; Strauss, E.C.; et al. Effector Mechanisms of the Autoimmune Syndrome in the Murine Model of Autoimmune Polyglandular Syndrome Type 1. J. Immunol. 2008, 181, 4072–4079.
- Evan, J.R.; Bozkurt, S.B.; Thomas, N.C.; Bagnato, F. Alemtuzumab for the treatment of multiple sclerosis. Expert Opin. Biol. Ther. 2018, 18, 323–334.
- Ruck, T.; Bittner, S.; Wiendl, H.; Meuth, S.G. Alemtuzumab in Multiple Sclerosis: Mechanism of Action and Beyond. Int. J. Mol. Sci. 2015, 16, 16414–16439.
- Agius, M.A.; Klodowska-Duda, G.; Maciejowski, M.; Potemkowski, A.; Eliezer, K.; Patra, K.; Wesley, J.; Madani, S.; Barron, G.; Katz, E.; et al. Safety and tolerability of inebilizumab (MEDI-551), an anti-CD19 monoclonal antibody, in patients with relapsing forms of multiple sclerosis: Results from a phase 1 randomised, placebo-controlled, escalating intravenous and subcutaneous dose study. Mult. Scler. J. 2019, 25, 235–245.
- Frampton, J.E. Inebilizumab: First Approval. Drugs 2020, 80, 1259–1264.
- Edwards, J.C.; Szczepanski, L.; Szechinski, J.; Filipowicz-Sosnowska, A. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N. Engl. J. Med. 2004, 350, 2572–2581.
- He, D.; Guo, R.; Zhang, F.; Zhang, C.; Dong, S.; Zhou, H. Rituximab for relapsing-remitting multiple sclerosis. Cochrane Database Syst. Rev. 2013, 2013, CD009130.
- McGinley, M.P.; Moss, B.; Cohen, J.A. Safety of monoclonal antibodies for the treatment of multiple sclerosis. Expert Opin. Drug Saf. 2017, 16, 89–100.
- Teng, Y.K.O.; Bruce, I.N.; Diamond, B.; Furie, R.A.; Van Vollenhoven, R.F.; Gordon, D.; Groark, J.; Henderson, R.B.; Oldham, M.; Tak, P.P. Phase III, multicentre, randomised, double-blind, placebo-controlled, 104-week study of subcutaneous belimumab administered in combination with rituximab in adults with systemic lupus erythematosus (SLE): BLISS-BELIEVE study protocol. BMJ Open 2019, 9, e025687.
- Cambridge, G.; Leandro, M.J.; Teodorescu, M.; Manson, J.; Rahman, A.; Isenberg, D.; Edwards, J.C. B cell depletion therapy in systemic lupus erythematosus: Effect on autoantibody and antimicrobial antibody profiles. Arthritis Rheum. 2006, 54, 3612–3622.
- Pescovitz, M.D.; Torgerson, T.R.; Ochs, H.D.; Ocheltree, E.; McGee, P.; Krause-Steinrauf, H.; Lachin, J.; Canniff, J.; Greenbaum, C.; Herold, K.C.; et al. Effect of rituximab on human in vivo antibody immune responses. J. Allergy Clin. Immunol. 2011, 128, 1295–1302.e5.
- Roberts, D.M.; Jones, R.; Smith, R.M.; Alberici, F.; Kumaratne, D.S.; Burns, S.; Jayne, D.R. Rituximab-associated hypogammaglobulinemia: Incidence, predictors and outcomes in patients with multi-system autoimmune disease. J. Autoimmun. 2015, 57, 60–65.
- Eming, D.; Nagel, A.; Wolff-Franke, S.; Podstawa, E. Rituximab Exerts a Dual Effect in Pemphigus Vulgaris. J. Investig. Dermatol. 2008, 128, 2850–2858.
- Stasi, R.; Del Poeta, G.; Stipa, E.; Evangelista, M.L.; Trawinska, M.M.; Cooper, N.; Amadori, S. Response to B-cell–depleting therapy with rituximab reverts the abnormalities of T-cell subsets in patients with idiopathic thrombocytopenic purpura. Blood 2007, 110, 2924–2930.
- Hoch, W.; McConville, J.; Helms, S.; Newsom-Davis, J.; Melms, A.; Vincent, A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat. Med. 2001, 7, 365–368.
- Sangwook, O.; Kevin, O.C.; Aimee, P. MuSK Chimeric Autoantibody Receptor (CAAR) T Cells for Antigen-specific Cellular Immunotherapy of Myasthenia Gravis (2769). Neurology 2020, 94, 2769.
- Ellebrecht, C.T.; Bhoj, V.G.; Nace, A.; Choi, E.J.; Mao, X.; Cho, M.J.; Di Zenzo, G.; Lanzavecchia, A.; Seykora, J.T.; Cotsarelis, G.; et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 2016, 353, 179–184.
- Bollmann, F.M. Rheumatic autoimmune diseases: Proposed elimination of autoreactive B-cells with magnetic nanoparticle-linked antigens. Med. Hypotheses 2012, 78, 479–481.
- Imura, Y.; Ando, M.; Kondo, T.; Ito, M.; Yoshimura, A. CD19-targeted CAR regulatory T cells suppress B cell pathology without GvHD. JCI Insight 2020, 5, e136185.
- Townsend, M.; Monroe, J.G.; Chan, A.C. B-cell targeted therapies in human autoimmune diseases: An updated perspective. Immunol. Rev. 2010, 237, 264–283.
- Vincent, F.B.; Morand, E.F.; Mackay, F. BAFF and innate immunity: New therapeutic targets for systemic lupus erythematosus. Immunol. Cell Biol. 2012, 90, 293–303.
- Musette, P.; Bouaziz, J.D. B Cell Modulation Strategies in Autoimmune Diseases: New Concepts. Front. Immunol. 2018, 9, 622.
- Jagessar, S.A.; Heijmans, N.; Bauer, J.; Blezer, E.; Laman, J.D.; Migone, T.-S.; Devalaraja, M.N.; Hart, B.A. T Antibodies Against Human BLyS and APRIL Attenuate EAE Development in Marmoset Monkeys. J. Neuroimmune Pharmacol. 2012, 7, 557–570.
- Mackay, F.; Schneider, P. Cracking the BAFF code. Nat. Rev. Immunol. 2009, 9, 491–502.
- Furie, R.; Rovin, B.H.; Houssiau, F.; Malvar, A.; Teng, Y.O.; Contreras, G.; Amoura, Z.; Yu, X.; Mok, C.-C.; Santiago, M.B.; et al. Two-Year, Randomized, Controlled Trial of Belimumab in Lupus Nephritis. N. Engl. J. Med. 2020, 383, 1117–1128.
- Kaegi, C.; Steiner, U.C.; Wuest, B.; Crowley, C.; Boyman, O. Systematic Review of Safety and Efficacy of Atacicept in Treating Immune-Mediated Disorders. Front. Immunol. 2020, 11, 433.
- Kalled, S.L.; Cutler, A.H.; Ferrant, J.L. Long-term anti-CD154 dosing in nephritic mice is required to maintain survival and inhibit mediators of renal fibrosis. Lupus 2001, 10, 9–22.
- Wang, X.; Huang, W.; Schiffer, L.E.; Mihara, M.; Akkerman, A.; Hiromatsu, K.; Davidson, A. Effects of anti-CD154 treatment on B cells in murine systemic lupus erythematosus. Arthritis Rheum. 2003, 48, 495–506.
- Li, Y.; Chen, F.; Putt, M.; Koo, Y.K.; Madaio, M.; Cambier, J.C.; Cohen, P.L.; Eisenberg, R.A. B Cell Depletion with Anti-CD79 mAbs Ameliorates Autoimmune Disease in MRL/lpr Mice1. J. Immunol. 2008, 181, 2961–2972.
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080.
- Gatumu, M.; Skarstein, K.; Papandile, A.; Browning, J.L.; Fava, R.A.; Bolstad, A.I. Blockade of lymphotoxin-beta receptor signaling reduces aspects of Sjögren syndrome in salivary glands of non-obese diabetic mice. Arthritis Res. Ther. 2009, 11, R24.