Stromal interacting molecule 1 (STIM1), a single transmembrane-spanning domain protein mainly residing in the endoplasmic reticulum (ER), is the unique ER Ca2+sensor deputed to ER Ca2+refilling. STIM1 was identified in 2005 by RNA interference-based screening of proteins with known signaling motifs. Molecularly, STIM1, residing into the organelle, senses luminal Ca2+ concentration by its N-terminus with a low affinity with an apparent dissociation constant (Kd) of ~0.2–0.6 mM and interferes with several plasma membrane ionic proteins. In this way, STIM1 coordinates the complex Ca2+ entry mechanism called SOCE deputed to ER Ca2+ recharge.
- Ca homeostasis
- lysosomes
- endoplasmic reticulum (ER)
- Ca-storing organelles
- amyotrophic lateral sclerosis (ALS)
- STIM1
1. Introduction
Stromal interacting molecule 1 (STIM1), a single transmembrane-spanning domain protein mainly residing in the endoplasmic reticulum (ER), is the unique ER Ca 2+ sensor deputed to ER Ca 2+ refilling [1]. Historically, STIM1 was identified in 2005 by RNA interference-based screening of proteins with known signaling motifs [2]. Molecularly, STIM1 residing into the organelle senses luminal Ca2+ concentration by its N-terminus with a low affinity with an apparent dissociation constant (Kd ) of ~0.2–0.6 mM [3]. This low affinity is due to the high Ca2+ levels within the organelle allowing the function of STIM1 as a unique ER Ca2+ sensor. When ER Ca2+ diminishes, STIM1 may diffuse to the plasma membrane regions where it interacts with the channel Orai1 through its C-terminal domain, after the unfolding of the EF-hand-sterile alpha motif (SAM). This interaction triggers a localized increase in cytoplasmic Ca2+ that induces SERCA pump activation to refill ER of Ca2+ . Two alternative splicing variants of STIM1 have been cloned: the long isoform named STIM1L with an extra 106 amino acids on its C-terminus, and the short 90-kDa STIM1 molecule named STIM1S. STIM1L is highly expressed in skeletal muscle as well as in several other tissues, including spleen, lungs, liver, brain and heart but not in the kidney. In human myotubes, STIM1L is expressed during myogenesis in the proximity to the PM, thus mediating a fast SOCE [4]. STIM1L displays a stronger propensity towards canonical transient receptor potential channel 1 (TRPC1) since it is more prone to open this channel [5]. Very recently, a neuronal splice variant named STIM1B has been cloned; it is selectively targeted to presynaptic ER and is useful to replenish synaptic vesicles after Ca2+ depletion [6]. On the other hand, STIM2 induces a weak activation of Orai1 [7] that minimally contributes to Ca2+ entry and prompts a limited Ca2+ -dependent NFAT1 activation [8]. In this respect, a functional coupling between STIM2 and STIM1 has been proposed. In fact, when ER Ca2+ stores are maximally depleted, STIM2 targets Orai1/STIM1 complex to ER-PM junctions promoting assembly of the channel with the AKAP79/calcineurin signaling complex that promotes NFAT1 activation [8]. However, STIM1 plays the major role in clustering and activating Orai1. Therefore, STIM1 activation governs ER Ca2+ refilling through store-operated calcium entry (SOCE), a ubiquitous plasma membrane mechanism first described by Jim Putney in 1986 [9]. SOCE provides Ca2+ signals to regulate critical cell functions in many tissues after the generation of inositol 1,4,5-triphosphate (IP 3) in response to receptor stimulation and the release of Ca2+ from the ER. The molecular components involved in the process remained uncharacterized for several years. A major advancement in the field came with the identification of the calcium release-activated calcium (CRAC) channel mediating the highly Ca2+ selective current (ICRAC ) in mast cells and T lymphocytes [10,11,12,13,14,15,16]. Therefore, Ca 2+ influx mediated by CRAC channels is called SOCE, because it is regulated by the filling state of ER Ca2+ stores. ICRAC is a non-voltage activated, inwardly rectifying current, highly selective for Ca2+. Accordingly, it displays a Ca2+:Na+ permeability ratio of 1000:1 [17,18]. Of note, the single-channel conductance of CRAC is small as compared with that of other plasma membrane channels, and it has been estimated in about 24 fS in Jurkat T cells [18].
Thanks to the effort of the scientific community, much is actually known about the regulatory mechanisms of SOCE (i.e., arrangement, dynamics, stoichiometry of SOCE molecular components) not only in non-excitable cells, where it is the primary Ca2+ influx pathway, but also in neuronal cells. However, once the role of STIM1 has been established, elucidating the other molecular components of SOCE proved to be a challenging endeavor for almost two decades. A number of proteins have been proposed to interact with STIM1, which are located both in the plasma membrane and at different intracellular levels. Some of these proteins may be considered as molecular partners of STIM1 and include Orai, TRPC channels, L-type voltage-dependent Ca 2+ channels, plasma membrane Ca2+ -ATPase (PMCA), exportin1, transportin1, SERCA2, SERCA3, etc. On the other hand, many modulators of SOCE, including the myelin basic protein Golli, and some scaffold proteins helping STIM1 to interact with its partners have also been described. A complex cooperation among these molecular components occurs in different parts of the cell to orchestrate intracellular Ca2+ homeostasis. Of note, this equilibrated cooperation may be altered during neurodegenerative disorders, thus causing neuronal death.
Molecularly, after store depletion, STIM1 interacts with the above-mentioned proteins, directly or indirectly through specific scaffold proteins. For instance, the SOCE-mediated cytosolic Ca2+ increase is primarily sustained by the direct activation of Orai1 by STIM1 and then by the inhibition of PMCA activity by STIM1-TMEM20 complex (see Section 4 for more details). When ER Ca2+ is refilled by SERCA [19], Orai1 and STIM1 dissociate and return to their resting state. This mechanism may be altered under pathological conditions through the dysregulation of both the expression and the function of STIM1 partners, thus impairing the outcomes of their interaction.
2. STIM1 Scaffold Proteins
STIM1 can modulate several ionic partners directly or through scaffold proteins enabling the Ca 2+ sensor to interact with multiple transporters ( Table 2 ). In this respect, the 10-transmembrane-spanning segment protein TMEM20 located at ER level has been considered the most important STIM1 protein mediator [71]. In fact, it has also been called the partner of stromal interaction molecule 1 (POST), since it is relevantly involved in the binding of STIM1 to plasma membrane proteins including Orai1, PMCA, SERCAs, Na+ /K+ ATPase, as well as nuclear proteins located at the nuclear envelope such as importins-β and exportins. At the plasma membrane level, POST-Orai1 binding is store depletion-independent. POST downregulation or overexpression does not substantially affect Orai1-mediated CRAC, thus suggesting the possibility that POST could modulate Orai1 activity in response to other physiological stimuli, independently from store depletion events. Interestingly, POST knockdown determines an increase in PMCA activity in store-depleted cells in which the STIM1-POST complex is bound to PMCA [71]. This suggests that the interaction between STIM1-POST and PMCA reduces the pump Ca 2+ extrusion, thus determining a sustained cytosolic Ca2+ elevation.
| STIM1 Modulator | Localization | Effect | References |
|---|---|---|---|
| TMEM20/POST | ER membrane; plasma membrane (minor fraction) |
Scaffold protein involved in the binding of STIM1 to plasma membrane proteins (i.e., Orai1, PMCA, SERCAs, Na+/K+ ATPase) and nuclear membrane proteins (importins and exportins) | [71] |
| SARAF (SOCE-associated regulatory factor) |
ER membrane | It protects cells from Ca2+ overfilling by promoting the Ca2+-dependent slow inactivation of CRAC channels after the interaction with STIM1 | [80,81] |
| Fragment P100 of polycystin-1 | ? | Reduction in SOCE via direct inhibition of STIM1 translocation | [83] |
| ERp57 | ER lumen | Negative modulation of SOCE via regulation of STIM1 oligomerization | [84] |
| Stanniocalcin 2 | Extracellular (secreted); ER lumen |
Negative regulation of SOCE | [86] |
| EB1 | Microtubules | It restricts STIM1 to ER-PM junctions, thus preventing excessive SOCE and ER Ca2+ overload | [89] |
| Golli | Cell body, nucleus and processes | By interacting with the C-terminal domain of STIM1, it negatively regulates the activity of SOCCs |
[90,91] |
For instance, suppressing TMEM20/POST expression through miR-150 determines an increase in intracellular Ca 2+ levels after TCR stimulation in CD8 + T cells. Since this is essential to induce the expression of activation-associated genes, miR-150-induced reduction in TMEM20 allows naïve CD8 + T cells to express anergy-inducing genes, such as Casitas B lineage lymphoma b (Cbl-b), Egr2 and p27. These findings suggest the central role of TCR-mediated intracellular Ca 2+ regulation in naïve CD8 + T cells [72].
Another important STIM1-interacting protein is the microtubular protein EB1, involved in the formation of STIM1-SERCA2A complex [56]. By interacting with this protein, STIM1 may recruit SERCA2A after Orai1 association thus replenishing the intracellular Ca 2+ store by the generation of an inward current creating a local microdomain. After the complete refilling, the complex dissociates, thus silencing the inward current. Upon store depletion, STIM1 becomes strongly bound to the POST-targeted molecules SERCA, PMCA and Na+/K+ ATPase, as well as to the nuclear transporters, importins-β and exportins. Store depletion-dependent STIM1 binding to SERCA2 [56] and some karyopherins [70] have been reported previously.
Very recently NCX1 has been identified as a new partner of STIM1 in mediating SOCE, whose activation in the reverse mode may be facilitated by the local increase of Na+ concentration due to the interaction between STIM1 and TRPC6 in primary cortical neurons (Tedeschi V, Sisalli MJ, Pannaccione A, Piccialli I, Molinaro P, Annunziato L, Secondo A. Na+/Ca2+ exchanger isoform 1 (NCX1) and canonical transient receptor potential channel 6 (TRPC6) are recruited by STIM1 to mediate Store-Operated Calcium Entry in primary cortical neurons. Cell Calcium 2022 Jan;101:102525).
3. STIM1 Partners in Neurodegenerative Diseases
The involvement of STIM1 (and its partners) in neuronal injury has been demonstrated in diverse acute and chronic degenerative conditions ( Figure 1 ). Increased STIM1 expression precedes cell death of cortical neurons in rats exposed to lateral head rotational injury [92]. Similarly, traumatic brain injury triggers elevation of STIM1 expression that contributes to apoptotic cells death by upregulating metabotropic glutamate receptor (mGluR)1-dependent Ca2+ signalling [93].
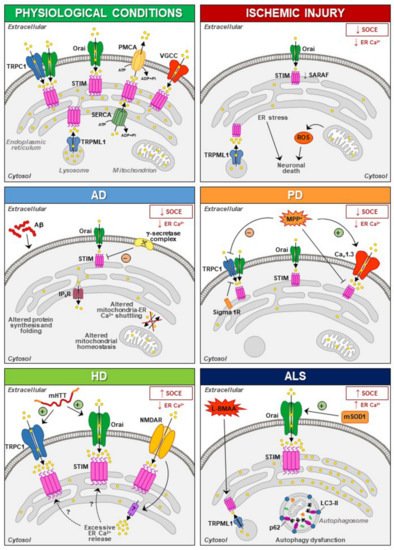
Thus, dysfunctions of STIM and NMDAR have been suggested to be involved in the pathogenesis of neurodegenerative diseases, although the mechanisms involved in the interplay between these two partners need to be further elucidated [96,97]. Overstimulation of NMDARs results in intracellular Ca2+ overload that underlies excitotoxic neuronal death in neurodegeneration occurring in brain ischemia, Huntington’s disease (HD) and Alzheimer’s disease (AD) [98,99,100,101]. In these contexts, targeting the mechanism that links NMDAR with STIM proteins might represent a promising neuroprotective strategy [95,102,103,104]. Accordingly, upregulating STIMs can protect against NMDA-induced dysfunctions of Ca 2+ homeostasis [94]. In addition to its role in the regulation of NMDAR-mediated excitotoxic injury, STIM1 has also been shown to be involved (directly or indirectly through its partners/effectors) in detrimental mechanisms specifically activated under diverse neurodegenerative conditions ( Figure 1 ).
These results strongly suggest that aberrant STIM1 levels contribute to the pathogenesis of FAD. Interestingly, downregulation of STIM1 may compromise its role in the regulation of Ca V 1.2, thus affecting neuronal activity in AD [51,52]. In fact, reduced STIM1 levels occurring in various AD cell models underlie defective Ca 2+ homeostasis and learning and memory impairment in a SOCE-independent manner. This includes regulation of ER refilling through mGluR1 and 5 [155,156] and of synaptic plasticity [157], as well as Ca 2+ overload through upregulation of L-type VGCCs and mitochondrial dysfunction [152]. A recent work has demonstrated that STIM1-deficient SH-SY5Y cells display reduced expression of the IP 3 receptors type 3 (IP 3R3) that is, in turn, responsible for the low mitochondrial Ca2+ concentration ([Ca2+ ]) and for the reduced efficiency of this organelle [158], which are common features in AD patients [159].
In PC12 cells treated with MMP+, SOCE blockade and STIM1 depletion reduce oxidative stress, prevent mitochondrial dysfunction and increase cell viability, supporting the hypothesis that SOCC through STIM1 underlies toxicity in this model [185]. In line with these findings is the recent evidence that Sigma 1 Receptors (σ1Rs) are involved in the inhibition of TRPC1-mediated Ca 2+ entry (by inhibiting STIM1 binding with TRPC1) in dopaminergic cells, whereby downregulation of σ1Rs expression or inhibition of σ1R activity substantially prevented MPP + -induced cell death by preserving Ca 2+ homeostasis, mitochondrial and ER function, and inhibiting apoptosis [186]. In particular, store depletion, especially in the presence of external Ca 2+ , causes the dissociation of σ1R-STIM1 complexes while facilitating STIM1-TRPC1-Orai1 interactions, which could also play a critical role in neuronal demise [186]. Nevertheless, the aforementioned data are in contrast with the observation that exposure to MPP + decreases TRPC1 expression, its interaction with STIM1 and Ca 2+ entry in SH-SY5Y cells [187], underscoring the need of additional experiments to clarify the role of STIM1 and its partners in PD pathogenesis.
4. Conclusions
SOCE elicits specific cytosolic Ca 2+ signals that are used by both excitable and non-excitable cells for regulating critical physiological processes. The ER Ca 2+ sensor STIM1 is responsible for SOCE machinery orchestration mainly through the interaction with Orai1 channel. However, several other STIM1 targets located in different cell compartments may serve to handle intracellular calcium concentration including plasmalemmal and intracellular channels (e.g., TRPC1, L-type voltage-dependent Ca2+ channels and TRPML1), and pumps (e.g., PMCA and SERCAs). Of note, the interaction between STIM1 and each of these ionic targets allows the regulation of both cytosolic and organellar Ca 2+ homeostasis. Moreover, through the modulation of STIM1 activity and interaction, many adaptors indirectly intervene in intracellular calcium homeostasis regulation.
Several lines of experimental evidence clearly demonstrate that Ca 2+ homeostasis and, most notably, STIM1 targets may be dysregulated in both acute and chronic neurodegenerative diseases. In fact, dysfunction of SOCE may contribute to the progression of several neurodegenerative diseases. For instance, SOCE activation appears to be neuroprotective in PD and AD, while during HD progression, neuroprotection could be achieved by reducing SOCE. With stroke injury being a multifactorial pathology dependent on the dysfunctional activity of several cell types, the role of SOCE in this neurological disease remains controversial. Therefore, dissection of the molecular components sustaining SOCE should be performed at the level of each specific cell type in order to identify the exact role played by each of them. This is true especially at the level of central nervous system (CNS) cells, where SOCE plays a dualistic role in the different forms of neurodegeneration. On the other hand, STIM1 interacts with other proteins not canonically involved in SOCE and this interaction may be altered during the neurodegenerative process. For instance, dysfunction in STIM1/TRPML1 interaction participates in ALS and stroke pathogenesis through organellar ionic dysfunction. This should be carefully taken into account since each element able to interact with STIM1 should be considered as a putative target for the development of new pharmacological entities.
Collectively, considering the relevance of Ca 2+ dyshomeostasis in neurodegeneration, the validation of new drugs toward STIM1 targets may result in successful treatment strategies for AD, PD, HD, ALS and stroke.
This entry is adapted from the peer-reviewed paper 10.3390/cells10102518