Ruben M. L. Colunga Biancatelli, Pavel Solopov and John D. Catravas
Heat shock protein 90 (HSP90) is an important chaperone that assists the late stage folding of several proteins involved in cell survival in response to environmental stressors. The inhibition of HSP90 is followed by a complex modulation of the proteome and the kinome, that has proved beneficial in cancer and various neurodegenerative diseases. Additionally, accumulating literature suggests that HSP90 may be a key target during the development of pulmonary fibrosis and that its inhibition could serve as a new and exciting therapeutic approach. We have summarized the current evidence about HSP90’s role in Idiopathic Pulmonary Fibrosis (IPF), the results from preclinical studies on its inhibition and the intracellular signaling pathways involved, in a recent review article (Review). In this Article entry, we will introduce the main findings discussed in the review and focus on its translation and possible significance in the era of the SARS-CoV-2 pandemic.
- Idiopathic pulmonary fibrosis
- heat shock proteins
- Proteome
- HSP90
- HSP90 inhibitors
- SARS-CoV-2
- COVID-19
- HSPome
- antifibrotic
HSP90 functions and isoforms
HSP90 is a highly expressed and conserved chaperone, described in diverse isoforms (e.g. α, β, mitochondrial), whose activity is necessary for the proper folding of many client proteins. Different isoform of HSP90 can assemble in heterogeneous or homologous dimers (HSP90αα, HSP90ββ or HSP90αβ), with specific chaperone profiles and affinity for protein partners [1][2]. Few studies have characterized both α and β isoforms behavior [3][4] during the fibrotic process. HSP90β has shown specific tubulin and microtubule-interacting properties playing a role in cell motility, migration and structural cell stability [5]. HSP90α expression is modulated by Signal Transducer and Activator of Transcription 1 (STAT1) and is involved in cell survival by modulating the caspase 3-depending apoptotic pathway [6]. However, more data is required for a better understanding of HSP90 isoforms, especially in order to develop inhibitors with lower or higher affinity.
HSP90 directs myofibroblast differentiation
Fibroblasts represent the central mediators of the fibrotic process as they promote cellular proliferation and deposition of extracellular matrix (ECM) in response to prolonged tissue injury and chronic inflammation [7]. Transforming growth factor-β (TGF-β) is the most important mediator of fibrogenesis, which upregulates and activates fibroblast phenotype and function, inducing fibroblast transdifferentiation [8]. TGF-β signaling is mediated by a complex subset of intracellular mediators divided into the non-Smad- and Smad-dependent signaling pathways [9][10]. HSP90 stabilizes the TGF-β receptor, modulates its signaling cascades and, similarly to TGF-β, directs myofibroblast differentiation.
Samples taken from patients with IPF revealed the presence of both HSP90α and HSP90β in the pulmonary interstitial tissue, whose expression was increased in areas rich in myofibroblasts and fibroblasts [3]. These fibroblasts localized HSP90 mainly in the nucleus and cytosol, and when treated with HSP90 inhibitors, displayed impaired proliferation, migration and differentiation [11]. Indeed, HSP90 administration increased the production of collagen by fibroblasts and the expression of α-Smooth Muscle Actin (α-SMA) by epithelial cells [4]. Thus, HSP90 promotes epithelial-to-mesenchymal transformation (EMT) of epithelial cells and differentiation of fibroblast into myofibroblasts, whereas its inhibition limits the expression of the aggressive phenotype of IPF fibroblasts.
HSP90 signaling in IPF proteomic studies
Proteomic profiling plays an important role in biomarker discovery. Proteomic analyses in IPF have been done on peripheral blood, bronchoalveolar lavage fluid (BALF) and lung tissue. While HSP90β-1 was found increased in peripheral blood of patients with IPF [12], both HSP90α and HSP90β were found increased in lung tissue [13][14]. HSP90 exerts distinct roles when expressed intracellularly, intranuclearly or released into the vascular compartment. Indeed, intracellular HSP90 acts as a chaperone, assisting the maturation, correcting the folding and when irreversibly damaged, promoting the degradation of several client proteins [15]. Differently, HSP90 can be released extracellularly in conditions of stress, promoting Antigen Presenting Cells (APC) maturation and, acting as “alarmins”, triggers the immune system response [16]. Thus, IPF displays high levels of HSP90 both intra- and extra-cellularly, which are required for both the augmented intracellular signaling -and subsequent elevated chaperone activity- and for the activation of the immune system in response to the chronic inflammation in the lung.
HSP90 inhibitors
HSP90 inhibitors modulate and reduce protein trafficking, restore proteostasis and diminish HSP90 chaperone activity, thus blocking the fibrotic process [17]. The number of HSP90 inhibitors has dramatically increased since the first molecule described in 1994 [18]. Geldanamycin is a natural product and a member of the benzoquinone annamycin family that has demonstrated anti-tumor and anti-proliferative characteristics, whose clinical implementation, however, was limited due to liver toxicity [19][20]. 17-N-allylamino-17-demethoxygeldanamycin (17-AAG) is an analog of geldanamycin with higher affinity for the ATP binding site of HSP90, and lower toxicity [21]. This inhibitor attenuated fibroblast activation and, also, fibroblast to myofibroblast transformation [11]. 17-AAG and 17-DMAG modulate ARAF, AKT, CDK4, MET, and PDK1 affecting principally protein kinase activity, as 34% of kinases are reduced and only 6% of them are upregulated [22]. Another HSP90 inhibitor, 1G6-D7, attenuated the severity of the fibrotic process in a murine model of bleomycin-induced pulmonary fibrosis by downregulating the levels of HSP90 [23].
AUY922, a second generation HSP90 inhibitor, successfully inhibits both isoforms of HSP90 (HSP90α and HSP90β) [24]. Our previous studies on AUY-922 have demonstrated its ability to depress phosphorylation of ERK, interfere with TGF-β signaling, and thereby, prevent the upregulation of ECM in Hydrochloric acid (HCl)- and nitrogen mustard (NM)-induced pulmonary fibrosis [25][26][27]. Dose and effects of various HSP90 inhibitors in in vivo models of pulmonary fibrosis are summarized in table 1 (Table 1). HSP90 inhibitors regulate TGF-β receptor stabilization, interfere with Smad and non-Smad (Raf, P-ERK) TGF-β signaling cascade, downregulate transcription factors, decrease EMT and reduce the production of pro-fibrotic mediators and ECM.
Table 1. HSP90 inhibitors in in vivo studies on Pulmonary Fibrosis.
| Study | Species | Etiology | HSP90 inhibitor | Dose | Time of administration | Dowregulated proteins |
| Sontake et al. [11] | mice | Bleomycin | 17-AAG | 15 mg/Kg | 1x/day for 3 weeks | Hydroxyproline, α-SMA, Ki67 |
| Sibinska et al. [3] | mice | Bleomycin | 17-DMAG | 10 mg/Kg | 1 every 2 days for 2 weeks | Collagen I, TGF-β, α-SMA |
| mice | Bleomycin | 17-DMAG | 25 mg/Kg | 1 every 2 days for 2 weeks | No significant changes | |
| Dong et al. [23] | mice | Bleomycin | 1G6-D7 | 1 mg/Kg | 1x/day for 3 weeks | HSP90α, IL-6, IL-4, Collagen I, Fibronectin, pERK, pAKT, p-p38 |
| Marinova et al. [25] | mice | Hydrochloric acid | AUY-922 | 1 mg/Kg | 2x/week for 2 weeks | pHSP90, TGFβ, pERK, Collagen I, Hydroxyproline, Fibronectin |
| mice | Hydrochloric acid | AUY-922 | 1 mg/Kg | 2x/week for 4 weeks | pHSP90, TGFβ, pERK, Collagen I, Fibronectin | |
| Solopov et al. [27] | mice | nitrogen mustard | AUY-922 | 1 mg/Kg | 2 x/week for 10 days | No significant changes |
| mice | nitrogen mustard | AUY-922 | 1 mg/Kg | 2 x/week for 30 days | pHSP90, pERK, Collagen I, Fibronectin, Elastin | |
| mice | nitrogen mustard | AUY-922 | 2 mg/Kg | 3 x/week for 30 days | pHSP90, pERK, Collagen I, Fibronectin, Elastin |
αSMA: α-Smooth Muscle Actin; TGFβ: Transforming growth factor-β; HSP90α: Heat Shock Protein 90 α; IL-6: Interleukin-6; IL-4: Interleukin-4; pERK: phospho- Extracellular-Signal-Regulated Kinase, AKT: Protein Kinase B; Ki67: cellular marker for proliferation, p-p38: phospho- protein 38.
HSP90 inhibition modulates the fibrotic process
HSP90 stabilizes Transforming Growth Factor-β (TGF- β) receptor [28], guarantee its proper signaling via Smad and non-Smad dependent pathways [29][30] and promote collagen secretion via interaction with cytosolic components of the secretory pathway [31]. HSP90 inhibition reduces ERK levels, involved in the non-Smad downstream of TGF-β [25][27], blocks TGF-β induced Smad phosphorylation [32] and lowers circulating levels of HSP90α [23] (Figure 1). HSP90 inhibitors modulate the expression of the proteome (119 peptides) [33] and the kinome (144 kinases) [34] preventing epithelial-to-mesenchymal transformation (EMT) and reducing the production of pro-fibrotic mediators and ECM. HSP90 inhibitors then, modulating the "HSPome", not only block the crucial intracellular signaling of TGF-β, but also modulate the inflammatory markers (via extracellular HSP90) related to IPF.
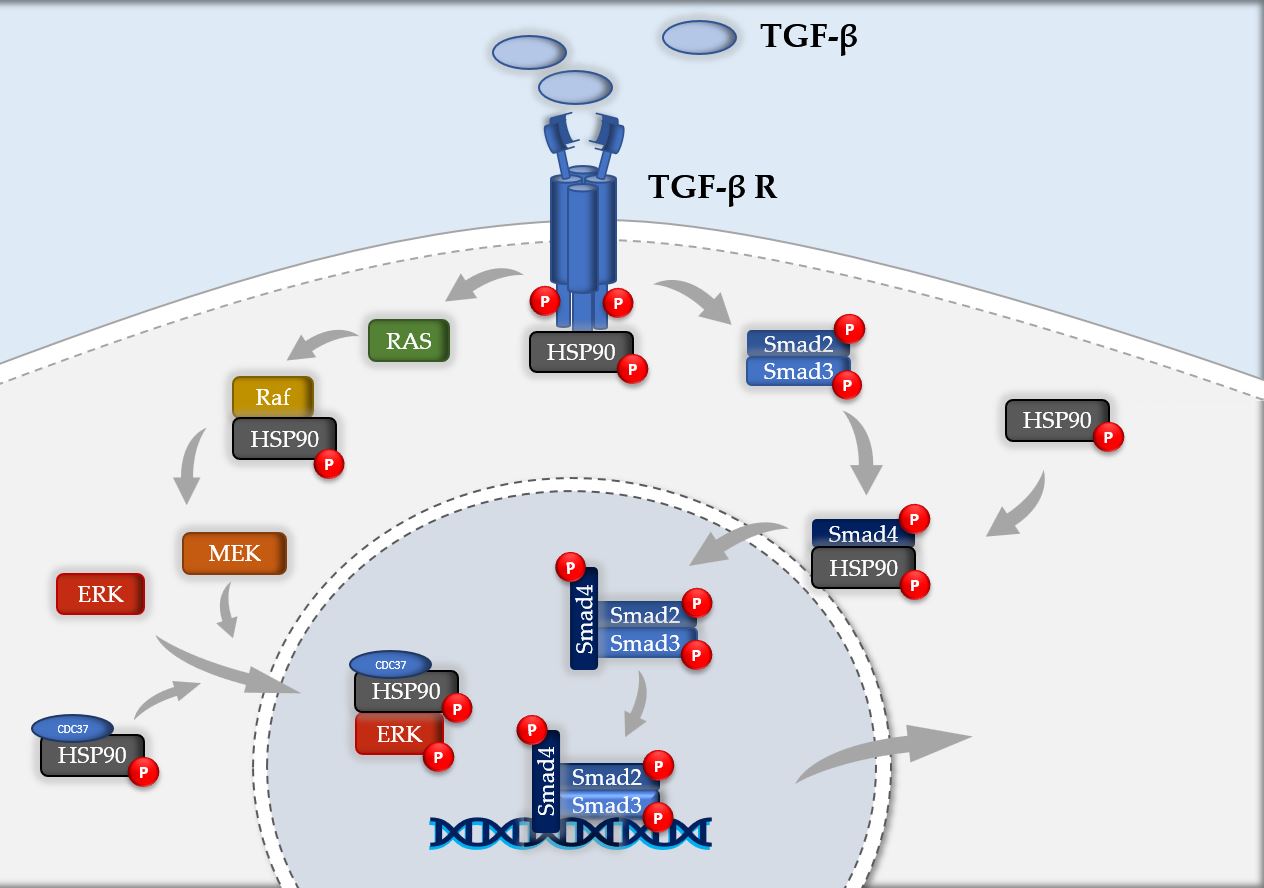
Figure 1. Schematic representation of HSP90 involvement in TGF-β signaling cascade. HSP90 plays a crucial role at various levels of the pathogenic pathway of IPF. It stabilizes TGF-β receptor, negatively regulates Raf, preserve ERK from degradation with its binding with HSP90-CDC37 complex and modulates nuclear localization of phospho-Smad4.
The need for new antifibrotic therapies in the era of SARS-CoV-2 Pandemic
Pirfenidone and Nintedanib are currently the only FDA approved drugs for IPF; their effect is limited to slowing disease progression [35] and, as of today, a cure is still missing. Furthermore, as several studies have reported increasing trends in mortality in IPF patients during the last years [36][37], the developing new therapeutical targets is still an unmet medical need. HSP90 inhibitors that are already in clinical trials for cancer, despite the promising results in animal models, require further experimentation for the definition of the optimal dosage and related safety in IPF.
Now more than ever, the development of new antifibrotic treatments, appears imperative because of the potential use in preventing the long-term complications from SARS-CoV-2 related disease (COVID-19). Indeed, COVID-19, similarly to ARDS, provokes long-lasting effects on the lung parenchyma and structure that can result in pleural thickening, atelectasis and pulmonary fibrosis [38][39], that, in patients with Middle-East Respiratory Syndrome (MERS), directly correlates with disease severity and Intensive Care Unit (ICU) length of stay [40]. It was recently proposed to initiate early antifibrotic therapies for COVID-19 patients with or without IPF [41]. Indeed, blocking TGF-β has been already suggested [42][43] since TGF-β is increased in lung sections of patients with COVID-19 [44]. Therefore, HSP90 inhibitors, capable of modulating the downstream cascades of TGF-β, have a strong translating potential for the treatment of COVID-19 patients; clearly, more preclinical and clinical studies are needed.
While few trials are under way to test the effectiveness of Pirfenidone and Nintedanib for the treatment of COVID-19 (NCT04282902, NCT04338802), even more need to focus on the development of new antifibrotic interventions with better efficacy.
Conclusion
In this Article Entry we briefly introduced the most significant results of HSP90 inhibition and modulation of the Proteome as potential therapies against the development of pulmonary fibrosis. We then translated their significance to the era of the SARS-CoV-2 pandemic.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21155286
References
- R S Gupta; Phylogenetic analysis of the 90 kD heat shock family of protein sequences and an examination of the relationship among animals, plants, and fungi species.. Molecular Biology and Evolution 1995, 1, 000, 10.1093/oxfordjournals.molbev.a040281.
- Kim Pepin; Fumitaka Momose; Nobushige Ishida; Kyosuke Nagata; Molecular Cloning of Horse Hsp90 cDNA and Its Comparative Analysis with Other Vertebrate Hsp90 Sequences.. Journal of Veterinary Medical Science 2000, 63, 115-124, 10.1292/jvms.63.115.
- Zaneta Sibinska; Xia Tian; Martina Korfei; Baktybek Kojonazarov; Janina Susanne Kolb; Walter Klepetko; Djuro Kosanovic; Malgorzata Wygrecka; Hossein A. Ghofrani; Norbert Weissmann; et al. Amplified canonical transforming growth factor-β signalling via heat shock protein 90 in pulmonary fibrosis.. European Respiratory Journal 2017, 49, 1501941, 10.1183/13993003.01941-2015.
- Pierre-Simon Bellaye; Chiko Shimbori; Toyoshi Yanagihara; David Carlson; Philip Hughes; Chandak Upagupta; Seidai Sato; Nolan Wheildon; Timothy Haystead; Kjetil Ask; et al. Synergistic role of HSP90α and HSP90β to promote myofibroblast persistence in lung fibrosis.. European Respiratory Journal 2018, 51, 1700386, 10.1183/13993003.00386-2017.
- Verónica Cambiazo; Mauricio Gonzalez; Cristián Isamit; Ricardo B Maccioni; The β-isoform of heat shock protein hsp-90 is structurally related with human microtubule-interacting protein Mip-90. FEBS Letters 1999, 457, 343-347, 10.1016/s0014-5793(99)01070-4.
- Yu-Ping Wu; Kazuko Kita; Nobuo Suzuki; Involvement of human heat shock protein 90? in nicotine-induced apoptosis. International Journal of Cancer 2002, 100, 37-42, 10.1002/ijc.10449.
- Ryan T. Kendall; Carol Feghali-Bostwick; Fibroblasts in fibrosis: novel roles and mediators. Frontiers in Pharmacology 2014, 5, 0, 10.3389/fphar.2014.00123.
- Anna Biernacka; Marcin Dobaczewski; Nikolaos G. Frangogiannis; TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196-202, 10.3109/08977194.2011.595714.
- Katharine H. Wrighton; Xia Lin; Xin-Hua Feng; Critical regulation of TGF signaling by Hsp90. Proceedings of the National Academy of Sciences 2008, 105, 9244-9249, 10.1073/pnas.0800163105.
- Kelly L Walton; Katharine E. Johnson; Craig A. Harrison; Targeting TGF-β Mediated SMAD Signaling for the Prevention of Fibrosis. Frontiers in Pharmacology 2017, 8, 0, 10.3389/fphar.2017.00461.
- Vishwaraj Sontake; Yunguan Wang; Rajesh K. Kasam; Debora Sinner; Geereddy B. Reddy; Anjaparavanda P. Naren; Francis X. McCormack; Eric S White; Anil Jegga; Satish K. Madala; et al. Hsp90 regulation of fibroblast activation in pulmonary fibrosis.. JCI Insight 2017, 2, e91454, 10.1172/jci.insight.91454.
- Jamie L. Todd; Megan L. Neely; Robert Overton; Katey Durham; Idu Gulati; Howard Huang; Jesse Roman; L. Kristin Newby; Kevin R. Flaherty; Richard Vinisko; et al. Peripheral blood proteomic profiling of idiopathic pulmonary fibrosis biomarkers in the multicentre IPF-PRO Registry.. Respiratory Research 2019, 20, 227-13, 10.1186/s12931-019-1190-z.
- YaQiong Tian; Hui Li; Yujuan Gao; Chuanmei Liu; Ting Qiu; Hongyan Wu; Mengshu Cao; Yingwei Zhang; Hui Ding; Jingyu Chen; et al. Quantitative proteomic characterization of lung tissue in idiopathic pulmonary fibrosis. Clinical Proteomics 2019, 16, 1-11, 10.1186/s12014-019-9226-4.
- Martina Korfei; Sigrid Schmitt; Clemens Ruppert; Ingrid Henneke; Philipp Markart; Benjamin Loeh; Poornima Mahavadi; Malgorzata Wygrecka; Walter Klepetko; Ludger Fink; et al. Comparative Proteomic Analysis of Lung Tissue from Patients with Idiopathic Pulmonary Fibrosis (IPF) and Lung Transplant Donor Lungs. Journal of Proteome Research 2011, 10, 2185-2205, 10.1021/pr1009355.
- Florian H. Schopf; Maximilian M. Biebl; Johannes Buchner; The HSP90 chaperone machinery. Nature Reviews Molecular Cell Biology 2017, 18, 345-360, 10.1038/nrm.2017.20.
- Camilo Colaco; Christopher R. Bailey; K. Barry Walker; James Keeble; Heat Shock Proteins: Stimulators of Innate and Acquired Immunity. BioMed Research International 2013, 2013, 1-11, 10.1155/2013/461230.
- Ruben M. L. Colunga Biancatelli; Pavel Solopov; Betsy Gregory; John Catravas; HSP90 Inhibition and Modulation of the Proteome: Therapeutical Implications for Idiopathic Pulmonary Fibrosis (IPF). IJMS 2020, 21, 5286, 10.3390/ijms21155286.
- Leonard M. Neckers; Brian Blagg; Timothy Haystead; Jane B. Trepel; Luke Whitesell; Didier Picard; Methods to validate Hsp90 inhibitor specificity, to identify off-target effects, and to rethink approaches for further clinical development. Cell Stress and Chaperones 2018, 23, 467-482, 10.1007/s12192-018-0877-2.
- Jeffrey G. Supko; Robert L. Hickman; Michael R. Grever; Louis Malspeis; Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemotherapy and Pharmacology 1994, 36, 305-315, 10.1007/bf00689048.
- Jörg Kreuter; Influence of the surface properties on nanoparticle-mediated transport of drugs to the brain.. Journal of Nanoscience and Nanotechnology 2004, 4, 484-8, 10.1166/jnn.2003.077.
- Gnana Prakasam Krishnamoorthy; Teresa Guida; Luigi Alfano; Elvira Avilla; Massimo Santoro; Francesca Carlomagno; Rosa Marina Melillo; Molecular Mechanism of 17-Allylamino-17-demethoxygeldanamycin (17-AAG)-induced AXL Receptor Tyrosine Kinase Degradation*. Journal of Biological Chemistry 2013, 288, 17481-17494, 10.1074/jbc.M112.439422.
- Kirti Sharma; R. Martin Vabulas; Boris Macek; Stefan Pinkert; Jürgen Cox; Matthias Mann; F.-Ulrich Hartl; Quantitative Proteomics Reveals That Hsp90 Inhibition Preferentially Targets Kinases and the DNA Damage Response. Molecular & Cellular Proteomics 2011, 11, 0, 10.1074/mcp.m111.014654.
- Hangming Dong; Lishan Luo; Mengchen Zou; Chaowen Huang; Xuan Wan; Yahui Hu; Yanqing Le; Haijin Zhao; Wei Li; Fei Zou; et al. Blockade of extracellular heat shock protein 90α by 1G6-D7 attenuates pulmonary fibrosis through inhibiting ERK signaling. American Journal of Physiology-Lung Cellular and Molecular Physiology 2017, 313, L1006-L1015, 10.1152/ajplung.00489.2016.
- Michael Rugaard Jensen; Joseph Schoepfer; Thomas Radimerski; Andrew J. Massey; Chantale T Guy; Josef Brueggen; Cornelia Quadt; Alan Buckler; Robert Cozens; Martin J Drysdale; et al. NVP-AUY922: a small molecule HSP90 inhibitor with potent antitumor activity in preclinical breast cancer models. Breast Cancer Research 2008, 10, R33-R33, 10.1186/bcr1996.
- Margarita Marinova; Pavel Solopov; Christiana Dimitropoulou; Ruben M. L. Colunga Biancatelli; John Catravas; Post-treatment with a heat shock protein 90 inhibitor prevents chronic lung injury and pulmonary fibrosis, following acute exposure of mice to HCl. Experimental Lung Research 2020, 46, 203-216, 10.1080/01902148.2020.1764148.
- Heat Shock Protein (HSP) 90 Inhibitors Prevent the Development of Nitrogen Mustard-Induced Chronic Lung Injury and Pulmonary Fibrosis in Mice . American Thoracic Society Journals. Retrieved 2020-8-6
- Pavel Solopov; Ruben M. L. Colunga Biancatelli; Margarita Marinova; Christiana Dimitropoulou; John Catravas; The HSP90 Inhibitor, AUY-922, Ameliorates the Development of Nitrogen Mustard-Induced Pulmonary Fibrosis and Lung Dysfunction in Mice. International Journal of Molecular Sciences 2020, 21, 4740, 10.3390/ijms21134740.
- P.-S. Bellaye; Olivier Burgy; Sebastien Causse; Carmen Garrido; Philippe Bonniaud; Heat shock proteins in fibrosis and wound healing: Good or evil?. Pharmacology & Therapeutics 2014, 143, 119-132, 10.1016/j.pharmthera.2014.02.009.
- Jeeyong Lee; You Sun An; Mi-Ra Kim; Ye-Ah Kim; Jin Kyung Lee; Chang Sun Hwang; Eunkyung Chung; In-Chul Park; Jae Youn Yi; Heat Shock Protein 90 Regulates Subcellular Localization of Smads in Mv1Lu Cells. Journal of Cellular Biochemistry 2015, 117, 230-238, 10.1002/jcb.25269.
- Tatiana Erazo; Ana Moreno; Gerard Ruiz-Babot; Arantza Rodríguez-Asiain; Nicholas A. Morrice; Josep Espadamala; Jose R. Bayascas; Néstor Gómez; Jose M. Lizcano; Canonical and Kinase Activity-Independent Mechanisms for Extracellular Signal-Regulated Kinase 5 (ERK5) Nuclear Translocation Require Dissociation of Hsp90 from the ERK5-Cdc37 Complex. Molecular and Cellular Biology 2013, 33, 1671-1686, 10.1128/MCB.01246-12.
- Madeline Y. Wong; Ngoc Duc Doan; Andrew S. DiChiara; Louis J. Papa; Jaime H. Cheah; Christian K. Soule; Nicki Watson; John D. Hulleman; Matthew D. Shoulders; A High-Throughput Assay for Collagen Secretion Suggests an Unanticipated Role for Hsp90 in Collagen Production. Biochemistry 2018, 57, 2814-2827, 10.1021/acs.biochem.8b00378.
- Hyunjin Noh; Hyun J Kim; Mi R Yu; Wan-Young Kim; Jin Kim; Jung H Ryu; Soon Hyo Kwon; Jin S Jeon; Dong C Han; Fuad Ziyadeh; et al. Heat shock protein 90 inhibitor attenuates renal fibrosis through degradation of transforming growth factor-β type II receptor. Laboratory Investigation 2012, 92, 1583-1596, 10.1038/labinvest.2012.127.
- Dongweon Song; Raghothama Chaerkady; Aik-Choon Tan; Elena García-García; Anuradha Nalli; Ana Suarez-Gauthier; Fernando Lopez-Rios; Xian Feng Zhang; Anna Solomon; Jeffrey Tong; et al. Antitumor activity and molecular effects of the novel heat shock protein 90 inhibitor, IPI-504, in pancreatic cancer. Molecular Cancer Therapeutics 2008, 7, 3275-3284, 10.1158/1535-7163.mct-08-0508.
- Armin Haupt; Gerard Joberty; Marcus Bantscheff; Holger Fröhlich; Henning Stehr; Michal-Ruth Schweiger; Axel Fischer; Martin Kerick; Stefan Thomas Börno; Andreas Dahl; et al. Hsp90 inhibition differentially destabilises MAP kinase and TGF-beta signalling components in cancer cells revealed by kinase-targeted chemoproteomics. BMC Cancer 2012, 12, 38-38, 10.1186/1471-2407-12-38.
- Ganesh Raghu; Bram Rochwerg; Yuan Zhang; Carlos Cuello-García; Arata Azuma; Juergen Behr; Jan Brozek; Harold R. Collard; William Cunningham; Sakae Homma; et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. American Journal of Respiratory and Critical Care Medicine 2015, 192, 1, 10.1164/rccm.201506-1063st.
- Dominic C. Marshall; Justin D. Salciccioli; Barry S. Shea; Praveen Akuthota; Trends in mortality from idiopathic pulmonary fibrosis in the European Union: an observational study of the WHO mortality database from 2001–2013. European Respiratory Journal 2018, 51, 1701603, 10.1183/13993003.01603-2017.
- Augustine S. Lee; Isabella Mira-Avendano; Jay H Ryu; Craig E. Daniels; The burden of idiopathic pulmonary fibrosis: An unmet public health need. Respiratory Medicine 2014, 108, 955-967, 10.1016/j.rmed.2014.03.015.
- Davide Chiumello; Silvia Coppola; Sara Froio; Miriam Gotti; What's Next After ARDS: Long-Term Outcomes. Respiratory Care 2016, 61, 689-699, 10.4187/respcare.04644.
- Giangaspare Mineo; F. Ciccarese; C. Modolon; M. P. Landini; M. Valentino; M. Zompatori; Post-ARDS pulmonary fibrosis in patients with H1N1 pneumonia: role of follow-up CT. La radiologia medica 2011, 117, 185-200, 10.1007/s11547-011-0740-3.
- Karuna M Das; Edward Y Lee; Rajvir Singh; Mushira A Enani; Khalid Al Dossari; Klaus Van Gorkom; Sven G Larsson; Ruth D Langer; Follow-up chest radiographic findings in patients with MERS-CoV after recovery. Indian Journal of Radiology and Imaging 2016, 27, 342-349, 10.4103/ijri.ijri_469_16.
- Peter M George; Athol U Wells; Gisli Jenkins; Pulmonary fibrosis and COVID-19: the potential role for antifibrotic therapy. The Lancet Respiratory Medicine 2020, 3, 2, 10.1016/s2213-2600(20)30225-3.
- Wanjun Chen; A potential treatment of COVID-19 with TGF-β blockade. International Journal of Biological Sciences 2019, 16, 1954-1955, 10.7150/ijbs.46891.
- Selectively targeting TGF-β with Trabedersen/OT-101 in treatment of evolving and mild ards in COVID-19 . Semantic scholar. Retrieved 2020-8-6
- Shih-Wen Li; T.-C. Yang; Lei Wan; Ying-Ju Lin; Fuu-Jen Tsai; Chien-Chen Lai; Cheng-Wen Lin; Correlation between TGF-β1 expression and proteomic profiling induced by severe acute respiratory syndrome coronavirus papain-like protease. PROTEOMICS 2012, 12, 3193-3205, 10.1002/pmic.201200225.
- Selectively targeting TGF-β with Trabedersen/OT-101 in treatment of evolving and mild ards in COVID-19 . Semantic scholar. Retrieved 2020-8-6
- Shih-Wen Li; T.-C. Yang; Lei Wan; Ying-Ju Lin; Fuu-Jen Tsai; Chien-Chen Lai; Cheng-Wen Lin; Correlation between TGF-β1 expression and proteomic profiling induced by severe acute respiratory syndrome coronavirus papain-like protease. PROTEOMICS 2012, 12, 3193-3205, 10.1002/pmic.201200225.