Reactive oxygen species (ROS) are important in regulating normal cellular processes whereas deregulated ROS leads to the development of a diseased state in humans including cancers. Several studies have been found to be marked with increased ROS production which activates pro-tumorigenic signaling, enhances cell survival and proliferation and drives DNA damage and genetic instability. However, higher ROS levels have been found to promote anti-tumorigenic signaling by initiating oxidative stress-induced tumor cell death. Tumor cells develop a mechanism where they adjust to the high ROS by expressing elevated levels of antioxidant proteins to detoxify them while maintaining pro-tumorigenic signaling and resistance to apoptosis. Therefore, ROS manipulation can be a potential target for cancer therapies as cancer cells present an altered redox balance in comparison to their normal counterparts.
- mitochondrial ROS
- oxidative stress
- cancer metabolism
- warburg effect
- tumor progression
- apoptosis
- autophagy
- NFκB pathway
- tumor adaptation
- drug resistance
- angiogenesis
- metastasis
- tumor targeting
1. Introduction
Reactive oxygen species (ROS), the partially reduced metabolites of oxygen that possess strong oxidizing capabilities, are deleterious to cells at high concentrations but at low concentrations, they serve complex signaling functions. Reactive oxygen species formed as byproducts of normal cell metabolism are needed for maintaining homeostasis and cellular signaling. Apart from cellular metabolism they are generated by specific plasma membrane oxidases in response to growth factors and cytokines and serve as secondary messengers in specific signaling pathways and play a role in regulating gene expression [1]. Cells have a defense system to maintain ROS at physiologically normal levels, i.e., enzymes called antioxidants, responsible for transforming free radicals into stable, less damaging molecules, the impairment of which may lead to a state of oxidative stress [2]. These oxygen scavenging pathways include conversion of O2− to H2O2 by superoxide dismutase (SOD), the action of catalase on H2O2 to produce H2O and O2, decomposition of H2O2 and LOOH by Glutathione peroxidase, and the reduction of H2O2 by Thioredoxin reduction cycle to produce H2O and also the exogenous detoxification of glutathione transferase [3]. Cancer cells are highly metabolically active and hypoxic cells, and due to massive growth and insufficient vascular irrigation tend to produce increased ROS, which damages DNA by diffusing through the mitochondrial membrane while also acting as signal-transducing messengers in many redox-sensitive molecular pathways involved in cell survival, therapeutic resistance, and progression [3]. Oxidative stress plays a major role in cancer hallmarks like angiogenesis, invasiveness, stemness, and metastatic ability, and hence, reducing oxidative stress with powerful antioxidants has been used as an important strategy for cancer prevention. Additionally, cancer cells develop mechanisms of keeping the increased oxidative stress in check. Therefore, some cancer therapeutic strategies also work by disrupting this check and making the cancer cells susceptible to apoptosis.
2. Source of Reactive Oxygen Species in Cancer Cells
2.1. Mitochondrial ROS
Mitochondria is one of the most prominent sources of reactive oxygen species within a cell which contribute to oxidative stress [4]. The electron transport chain located on the inner mitochondrial membrane generates the majority of mitochondrial ROS during the process of oxidative phosphorylation (OXPHOS). Leakage of electrons at complex I and complex III from ETC leads to a partial reduction of oxygen to form superoxide which undergoes spontaneous dismutation to hydrogen peroxide, both of which are collectively considered as mitochondrial ROS [5]. Endogenous modulators such as NO and Ca2+ have been observed to regulate the production of mtROS by regulating the metabolic states of mitochondria. The mitochondrial Ca2+ levels increase the rate of electron flow in the ETC and thus decrease mtROS generation [6]. However mitochondrial Ca2+ overload increases mtROS production [7]. STAT3, a transcription factor that regulates gene expression in response to cytokines interleukin (IL)-6 and IL-10, also modulates the activity of the ETC [8][9]. Hence a decrease in expression of STAT3 may be correlated to increasing mtROS at complex I [8]. TNF-α that causes the shedding of TNF-α receptor-1 reducing the severity of microvascular inflammation, has been found to induce a calcium-dependent increase in mt ROS [10]. Studies have shown that many ROS-producing enzymes, like NADPH oxidase, xanthine oxidase, and uncoupled eNOS, can stimulate mtROS production in a process called “ROS-induced ROS” [11][12][13]. Another transcription factor hypoxia-inducible factor 1α (HIF-1α) also plays a prominent role in bringing about a reduction in ROS by a number of mechanisms including induction of pyruvate dehydrogenase kinase 1 (PKD1), which shunts pyruvate away from the mitochondria; triggering mitochondrial selective autophagy; and induction of microRNA-210 blocking OXPHOS [14]. Low levels of mtROS regulate the stability of HIF-1α leading to hypoxia adaptation while moderate levels of mtROS have been found to regulate the production of proinflammatory cytokines by directly activating the inflammasome and mitogen-activated protein kinase (MAPK). However, high levels of mtROS are capable of inducing apoptosis by oxidation of the mitochondrial pores and autophagy by the oxidation of autophagy-specific gene 4 (ATG4) [5]. Depending on the tumor cell microenvironment, the c-Myc gene controls apoptosis by inducing aerobic glycolysis and/or OXPHOS which is required for the activation of certain tumor suppressor proteins, such as Bax and Bak [15][16][17].
Mitochondria also play an important role in the loss of caveolin 1 (cav-1) in the tumor-associated fibroblast compartment, which is related to the early tumor recurrence, metastasis, tamoxifen-resistance, and aggravated increase in tumor growth [2]. Cav-1 loss induces autophagy and mitophagy, [18] driving the “Reverse Warburg Effect” by a feed-forward mechanism. This onset of inflammation, autophagy, mitophagy, and aerobic glycolysis in the tumor microenvironment is triggered by activation of the transcription factors NFκB and HIF-1α [19][20]. Mitochondria-generated ROS plays an important role in cell proliferation and quiescence. The pro- or anti-tumorigenic signaling is controlled by a mitochondrial ROS switch of the antioxidant SOD2/MnSOD [21]. Cell proliferation is favored by decreased SOD2/MnSOD activity resulting in increased O2− production whereas proliferating cells transit into quiescence when SOD/MnSOD activity increases resulting in increased H2O2 activity [22]. Inactivation of mitochondrial antioxidant responses like the Thioredoxin reductase (TrxR); which causes reduction of oxidized Trx to produce reduced Trx that reacts with ROS, contributes to increased oxidative stress in cancer cells.
Studies have shown that the cellular redox status is impacted by the recruitment of mitochondria by the expression of hTERT. This observation is supported by the presence of hTERT in the mitochondria and since mitochondrial-dependent apoptosis in target cells can be carried about by introducing hTERT inhibitors [23].
2.2. Role of Warburg Effect in ROS
The increased metabolic requirements of the cancer cells are met by upregulation of glucose transport and metabolism irrespective of oxygen supply [24]. There is also some evidence that cancer cells decrease mitochondrial respiration in the presence of oxygen, which suppresses apoptosis [25]. Under hypoxic conditions, the accelerated metabolism produces ROS in cancer cells that is countered by the increased NADPH which is met by the upregulated glycolysis [26][27]. NADPH is an essential cofactor for replenishing reduced glutathione (GSH) which is a critical antioxidant. Therefore, not only are cancer cells’ multiple urgent requirements catered to but cancer cell oxidative stress is also controlled by the Warburg effect [8]. Tumor cells have been reported to switch between the isoforms of pyruvate kinase, used in the last steps of glycolysis [28]. PKM2 the isoform found in high levels in tumor cells is slower and leads to the accumulation of PEP which in turn activates PPP by feedback inhibition of the glycolytic enzyme triosephosphate isomerase (TPI). This produces more NADPH which reduces ROS and further amplifies the inhibitory effect of PKM2 [26][27], Therefore ROS and PKM2 form a negative feedback loop to maintain ROS in a tolerable and functional range. The ROS-regulated gene, hypoxia-inducible factors (HIF-1α) regulates hypoxia-associated genes, some of which are associated with the Warburg effect and its accompanying pathways and hence, are a target of cancer therapies. PKM2 has been found to be the prolyl hydroxylases (PHDs)-induced coactivator for HIF-1α [8][29]. HIF-1α also regulates the MYC proto-oncogene which produces MYC protein [30] that regulates genes participating in energy generation and cell growth and proliferation. HIF-1α and MYC activate hexokinase 2 (HK-II) and pyruvate dehydrogenase kinase 1 (PDK1), which inhibits TCA and increases conversion of glucose to lactate [31]. Glucose transporter 1 (GLUT1) and lactate dehydrogenase A (LDHA) are also activated by HIF1 and MYC independently, resulting in increased glucose influx and higher glycolytic rates [13]. Warburg effect increases steady-state ROS condition in cancer cells by producing lactate that is extruded through monocarboxylate transporters to the microenvironment of cancer cells which has no antioxidant properties in contrast to pyruvate, citrate, malate, and oxaloacetate together with the reducing equivalents (NADH.H+) which are antioxidant intermediates. This increased oxidative stress in cancer cells is stopped from reaching cytotoxic levels by some antioxidant effects exerted by hexokinase II (HK II) and NADPH.H+ produced through HMP shunt. Latest studies show tumor cells have the capability to carry about both glycolytic and oxidative phosphorylation (OXPHOS) metabolism which makes them resistant to oxidative stress through enhanced antioxidant response and increased detoxification capacity [32]. The changes related to energy metabolism may be correlated to the expression of certain p53 downstream genes regulated by it, including SCO2, TIGAR, and the p53 inducible gene 3 (PIG3) [33][34][35].
2.3. NADPH Oxidase, Cox, and Xanthine Oxidase Produce ROS
The NADPH oxidases NOX catalytic subunit carries about the transfer of electrons from NADPH to the molecular oxygen producing ROS as their primary function [36]. The other oxidases like the mitochondrial electron transport chain produce superoxide (O2•–) as a by-product of another oxidative reaction. Furthermore, xanthine dehydrogenase gets converted to xanthine oxidase which is a dysfunctional variant of the parent enzyme which generates uncoupled eNOS. The kinetics of ROS formation and the nature of the ROS produced are different in the four nonphagocytic NADPH oxidase isoforms. Electrons transfer across the biological membranes through NOX and produce O2− which gets rapidly converted to H2O2 [17]. This H2O2 after diffusing across the membrane can affect multiple cellular signaling events. Increased NOX-derived ROS in cancer cells affects two major characteristics of cancer progression, i.e., stimulation of cell survival and genomic instability. H2O2 activates MAPK signaling, neutrophil phagocytosis, apoptosis, cellular senescence, and cell growth. It also plays a significant role in oxygen sensing and under hypoxic conditions, it stimulates the release of hypoxia-inducible factor (HIF-1α) and then vascular endothelial growth factor (VEGF) thus promoting angiogenesis [37].
2.4. ER Stress Leads to ROS
In the endoplasmic reticulum, the catalytic processes of oxidoreductase Ero1 and NADPH oxidase (NOX) produce ROS. The major source of cellular ROS is the oxidative protein folding carried about by Ero1 which uses the oxidative power of molecular oxygen to initiate redox relays which ultimately leads to disulfide bond formation in the newly folded proteins. The luminal H2O2 arising from Ero1 and NOX are scavenged by ER peroxidases, such as peroxiredoxin 4 (Prx4), as well as the glutathione peroxidases GPx7 and GPx8 and thereby prevent H2O2 leakage from the ER [38]. The accumulation of unfolded proteins, i.e., persistent ER stress leads to redox-amplified imbalances in the Ero1/PDI electron flow increasing production of ROS at the ER which can be counteracted by an influx of reduced glutathione (GSH) [39]. The production of ROS activates the unfolded protein response (UPR) inactivating the sulfhydration of protein tyrosine phosphatase 1B (PTP1B). This results in increased phosphorylation of PKR-like endoplasmic reticulum kinase (PERK) thereby activating it. PERK plays an important role in restoring cellular homeostasis by regulating a switching mechanism between autophagy and apoptosis [40]. ROS-mediated ER stress also signals activation of Nrf2 antioxidant response, thereby increasing stress resistance and lifespan [41][42]. ROS can leak through the ER through the aquaporin 8, the ER ROS pore. Similarly, Peroxisomal ROS production can leak into the cytosol, and lead to oxidation of important signaling molecules like the NF-kB and PTEN (Figure 1).
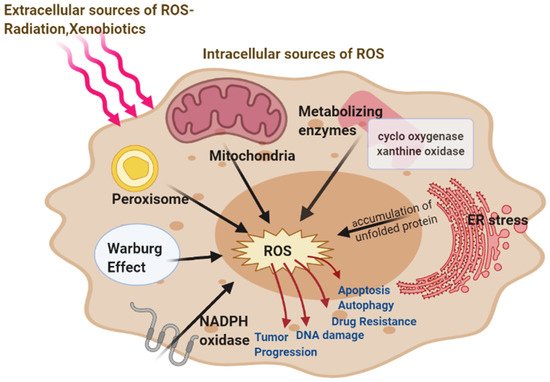
Figure 1. Major intracellular Sources of ROS-mitochondria, peroxisome, endoplasmic reticulum (ER) stress, nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase, metabolizing enzymes, and extracellular (Radiations, Xenobiotics) sources of reactive oxygen species (ROS) generation. ROS involved in cancer resulting in the development and progression of the disease.
This entry is adapted from the peer-reviewed paper 10.3390/antiox10050642
References
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Cell. Mol. Physiol. 2000, 279, L1005–L1028.
- Halliwell, B. Biochemistry of oxidative stress. Biochem. Soc. Trans. 2007, 35, 1147–1150.
- Vera-Ramirez, L.; Ramirez-Tortosa, M.; Perez-Lopez, P.; Granados-Principal, S.; Battino, M.; Quiles, J.L. Long-term effects of systemic cancer treatment on DNA oxidative damage: The potential for targeted therapies. Cancer Lett. 2012, 327, 134–141.
- Starkov, A.A. The Role of Mitochondria in Reactive Oxygen Species Metabolism and Signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52.
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.-F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19.
- Robert, F.F. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front. Biosci. 2009, 14, 1197–1218.
- Peng, T.-I.; Jou, M.-J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 2010, 1201, 183–188.
- Handy, D.E.; Loscalzo, J. Redox Regulation of Mitochondrial Function. Antioxid. Redox Signal. 2012, 16, 1323–1367.
- Wegrzyn, J.; Potla, R.; Chwae, Y.-J.; Sepuri, N.B.V.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of Mitochondrial Stat3 in Cellular Respiration. Science 2009, 323, 793–797.
- Dada, L.A.; Sznajder, J.I. Mitochondrial Ca2+ and ROS Take Center Stage to Orchestrate TNF-α-Mediated Inflammatory Responses. J. Clin. Investig. 2011, 121, 1683–1685.
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular Mechanisms of Angiotensin II–Mediated Mitochondrial Dysfunction. Circ. Res. 2008, 102, 488–496.
- Baudry, N.; Laemmel, E.; Vicaut, E. In vivo reactive oxygen species production induced by ischemia in muscle arterioles of mice: Involvement of xanthine oxidase and mitochondria. Am. J. Physiol. Circ. Physiol. 2008, 294, H821–H828.
- Ceylan-Isik, A.F.; Guo, K.K.; Carlson, E.C.; Privratsky, J.R.; Liao, S.-J.; Cai, L.; Chen, A.F.; Ren, J. Metallothionein Abrogates GTP Cyclohydrolase I Inhibition–Induced Cardiac Contractile and Morphological Defects. Hypertension 2009, 53, 1023–1031.
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Biophys. Acta (BBA) Bioenerg. 2011, 1813, 1263–1268.
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787.
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nat. Cell Biol. 2009, 458, 762–765.
- Tomiyama, A.; Serizawa, S.; Tachibana, K.; Sakurada, K.; Samejima, H.; Kuchino, Y.; Kitanaka, C. Critical Role for Mitochondrial Oxidative Phosphorylation in the Activation of Tumor Suppressors Bax and Bak. J. Natl. Cancer Inst. 2006, 98, 1462–1473.
- Martinez-Outschoorn, U.E.; Pavlides, S.; Whitaker-Menezes, D.; Daumer, K.M.; Milliman, J.N.; Chiavarina, B.; Migneco, G.; Witkiewicz, A.K.; Martinez-Cantarin, M.P.; Flomenberg, N.; et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: Implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle 2010, 9, 2423–2433.
- Outschoorn, U.E.; Lin, Z.; Trimmer, C.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect. Cell Cycle 2011, 10, 2504–2520.
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.B.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.M.; Lin, Z.; Witkiewicz, A.K.; et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution. Cell Cycle 2010, 9, 3276–3296.
- Sarsour, E.H.; Venkataraman, S.; Kalen, A.L.; Oberley, L.W.; Goswami, P.C. Manganese superoxide dismutase activity regulates transitions between quiescent and proliferative growth. Aging Cell 2008, 7, 405–417.
- Wang, M.; Kirk, J.S.; Venkataraman, S.; Domann, F.E.; Zhang, H.J.; Schafer, F.Q.; Flanagan, S.W.; Weydert, C.J.; Spitz, D.R.; Buettner, G.R.; et al. Manganese superoxide dismutase suppresses hypoxic induction of hypoxia-inducible factor-1α and vascular endothelial growth factor. Oncogene 2005, 24, 8154–8166.
- Indran, I.R.; Hande, M.P.; Pervaiz, S. Tumor cell redox state and mitochondria at the center of the non-canonical activity of telomerase reverse transcriptase. Mol. Asp. Med. 2010, 31, 21–28.
- Chen, X.; Qian, Y.; Wu, S. The Warburg effect: Evolving interpretations of an established concept. Free Radic. Biol. Med. 2015, 79, 253–263.
- Ruckenstuhl, C.; Büttner, S.; Carmona-Gutierrez, D.; Eisenberg, T.; Kroemer, G.; Sigrist, S.J.; Fröhlich, K.-U.; Madeo, F. The Warburg Effect Suppresses Oxidative Stress Induced Apoptosis in a Yeast Model for Cancer. PLoS ONE 2009, 4, e4592.
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.-K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of Pyruvate Kinase M2 by Reactive Oxygen Species Contributes to Cellular Antioxidant Responses. Science 2011, 334, 1278–1283.
- Hamanaka, R.B.; Chandel, N.S. Warburg Effect and Redox Balance. Science 2011, 334, 1219–1220.
- Sugiyama, T.; Taniguchi, K.; Matsuhashi, N.; Tajirika, T.; Futamura, M.; Takai, T.; Akao, Y.; Yoshida, K. MiR-133b inhibits growth of human gastric cancer cells by silencing pyruvate kinase muscle-splicer polypyrimidine tract-binding protein 1. Cancer Sci. 2016, 107, 1767–1775.
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate Kinase M2 Is a PHD3-Stimulated Coactivator for Hypoxia-Inducible Factor 1. Cell 2011, 145, 732–744.
- Liu, J.; Levens, D. Making Myc. Curr. Top. Microbiol. Immunol. 2006, 302, 1–32.
- Dang, C.V.; Kim, J.-W.; Gao, P.; Yustein, J. The interplay between MYC and HIF in cancer. Nat. Rev. Cancer 2008, 8, 51–56.
- Gentric, G.; Mieulet, V.; Mechta-Grigoriou, F. Heterogeneity in Cancer Metabolism: New Concepts in an Old Field. Antioxid. Redox Signal. 2017, 26, 462–485.
- Schmidt, M.W. Element Partitioning: The Role of Melt Structure and Composition. Science 2006, 312, 1646–1650.
- Won, K.Y.; Lim, S.-J.; Kim, G.Y.; Kim, Y.W.; Han, S.-A.; Song, J.Y.; Lee, D.-K. Regulatory role of p53 in cancer metabolism via SCO2 and TIGAR in human breast cancer. Hum. Pathol. 2012, 43, 221–228.
- Kotsinas, A.; Aggarwal, V.; Tan, E.-J.; Levy, B.; Gorgoulis, V.G. PIG3: A novel link between oxidative stress and DNA damage response in cancer. Cancer Lett. 2012, 327, 97–102.
- Bedard, K.; Krause, K.H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313.
- Sedeek, M.; Nasrallah, R.; Touyz, R.M.; Hébert, R.L. NADPH Oxidases, Reactive Oxygen Species, and the Kidney: Friend and Foe. J. Am. Soc. Nephrol. 2013, 24, 1512–1518.
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018, 9, 331.
- Chakravarthi, S.; Bulleid, N.J. Glutathione Is Required to Regulate the Formation of Native Disulfide Bonds within Proteins Entering the Secretory Pathway. J. Biol. Chem. 2004, 279, 39872–39879.
- Liu, Z.; Lv, Y.Z.; Zhao, N.; Guan, G.; Wang, J. Protein kinase R-like ER kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell Death Dis. 2015, 6, e1822.
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426.
- Hourihan, J.M.; Mazzeo, L.E.M.; Fernández-Cárdenas, L.P.; Blackwell, T.K. Cysteine Sulfenylation Directs IRE-1 to Activate the SKN-1/Nrf2 Antioxidant Response. Mol. Cell 2016, 63, 553–566.