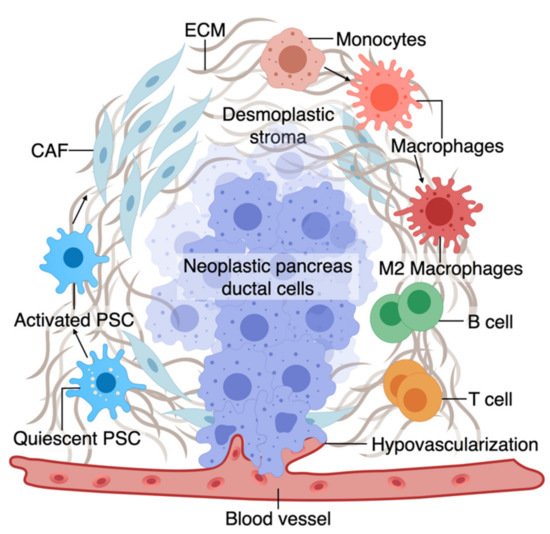
The TME has an active role in tumor progression, immune evasion, and drug response in PDAC [
70,
71]. Non-cellular components of TME of pancreas tumors are composed of ECM and dense fibrous tissue, regarded as “desmoplasia” (
Figure 1) [
72]. The dense ECM is mainly composed of matrix proteins such as collagen secreted by the cellular components of TME such as fibroblasts and pancreatic stellate cells (PSCs) [
73]. PSCs can reduce cancer cell death upon chemotherapy induction by releasing soluble factors or activating stemness signaling pathways in cancer cells [
15]. Several other immune cell types are present in the TME, among which macrophages are the most abundant that gauge both the innate and adaptive immune responses against the tumor [
73]. Similar to PSCs, macrophages in interaction with cancer cells can shift towards a pro-tumorigenic phenotype, which leads to increased cancer cell stemness and growth [
74]. Macrophages are also crucial in rendering chemoresistance to conventional chemotherapeutics for PDAC [
75]. Pancreatic epithelial ducts secrete alkali in the epical side and create an alkaline pH in the microenvironment to prevent the breakdown of secreted pre-enzymes in the normal pancreas before reaching the small bowel lumen [
76]. In PDAC, dysregulation of ion channel transporters (i.e., Ca
2+ and K
+ channels) and the tumor hypovascularization contribute to the acidification of the microenvironment, which further promotes cancerous characteristics in the epithelium (e.g., selection of EMT phenotypes) while shifting the stromal cells (i.e., PSC and macrophages) towards protumorigenic phenotypes [
76,
77]. While there are numerous other cell types in the TME (e.g., T cells, B cells, Dendritic cells), we focus on collagen-producing cells and macrophages as two major components of the TME in PDAC. In the following sections, we highlight the characteristics and plasticity of these cell types within the tumor area before discussing the opportunities provided by OOCs to model TME in PDAC.
Figure 1. A. The tumor microenvironment of PDAC is composed of interactions among different cell types. At an early stage, interactions of transforming neoplastic ductal cells with pancreatic stellate cells, fibroblasts, and macrophages result in initiating a fibroinflammatory process and subsequent involvement of an adaptive immune response, including recruitment of T cells. As the tumor progresses, cancer cells invade the blood vessels and occupy the vessel lumen, referred to as endothelial ablation. Endothelial ablation and a desmoplastic stroma, accompanied by a suppressive immune environment, reduce drug delivery and chemoresistance at the site.
2.1. Macrophages and Fibroblasts in PDAC
Macrophages in the pancreatic tissue are immune cells that can arise from the embryonic precursors traced back to the extraembryonic yolk or infiltrating monocytes from myeloid precursors in the bone marrow [
78,
79,
80,
81,
82,
83]. The tissue macrophages can change their function (polarization) in response to surrounding signals. While macrophages were traditionally classified into M1 and M2 subtypes, mounting evidence supports the presence of a wide phenotypic spectrum between M1 with stronger killing properties to M2 that can contribute to a smoldering chronic inflammatory state in cancer [
83,
84,
85,
86,
87]. M1 macrophages, through the production of nitrogen and oxygen derivatives, possess anti-tumorigenic ability by identifying and destroying cancer cells through phagocytosis [
84].
In contrast, M2 macrophages can promote tumor growth via multiple mechanisms, as discussed in detail elsewhere [
84,
88]. While M1 macrophages could be predominant in the TME during the early stages of cancer formation, the M2-type phenotype becomes more abundant as the tumor progresses. In fact, tumor-associated macrophages are more similar to M2-types and predict poor survival [
88,
89,
90]. Macrophage polarization or recruitment within the pancreas tissue occurs via crosstalk with cancerous epithelium and other components of the TME. In line with the growing literature on the crosstalk between macrophages and cancerous epithelium in PDAC, we also showed that early carcinogenesis signaling in the pancreatic epithelium could shift macrophages towards M2-like cells and that polarized macrophages could further promote cancer formation via induction of inflammatory signaling [
9,
73].
The major source of matrix deposition in the TME is cancer-associated fibroblasts (CAFs) that can arise from the PSCs, the resident mesenchymal cells in the pancreas [
91,
92,
93]. Cancer cells activate resident PSCs, which differentiate into CAFs and secrete matrix proteins such as collagen, contributing to the dense ECM [
89]. Activated PSCs also secrete factors to induce tumor growth, progression, and metastasis [
90,
91,
94,
95].
While the dense ECM has been traditionally considered to help tumor progression, recent studies support a more complex role for ECM’s contribution to PDAC progression, which could be stage and context-dependent [
96,
97]. However, at a late stage, the ECM contributes to tumor chemoresistance via multiple mechanisms such as cancer cell sensitivity, drug cytotoxicity, and reduced drug delivery [
98,
99,
100]. Tumor fibroblasts are associated with poor drug response and disease survival partly by offsetting chemotherapy-induced apoptosis via soluble factors or activating stemness signaling pathways in the cancer cells [
101,
102,
103,
104,
105]. Similar to macrophages, pancreatic fibroblasts could be phenotypically plastic and dynamic in response to the surrounding tumor stimuli [
106,
107]. Although continuous matrix deposition turns on the signaling pathways to boost the malignant phenotype, more desmoplasia contributes to tumor progression and drug resistance [
108,
109,
110]. While the role of stroma in promoting resistance in drug response is well accepted, recent animal studies show conflicting roles of stroma in tumor formation in PDAC. Protective effects of stroma were suggested by increased tumor aggression upon stroma reduction in a mouse model where Sonic hedgehog (Shh), a soluble ligand that drives the formation of desmoplastic stroma, was deleted [
111].
2.2. TME on Chip Models of PDAC
A biomimetic ductal TME on a chip, where ductal pancreatic cancer epithelium cells were surrounded by collagen matrix in the chip, recapitulated the histopathology of PDAC [
112]. The tumor heterogeneity was reconstituted using pancreatic cancer cells from GEMM carrying KRAS, CDKN2A, and TP53 mutations, key driver mutations of human PDAC. This model revealed the complex interactions between cancerous epithelial cells in PDAC, leading them to be more aggressive and invasive [
112].
PDAC is highly metastatic even at an early stage and escapes into the blood circulation [113,114]. In a biomimetic OOCs based model of PDAC, endothelial ablation was seen where PDAC cells invaded the matrix toward the endothelial lumen, wrapped around the blood vessel, spread along the length, and finally invaded the vessel and occupied the vessel lumen [66]. This finding of endothelial ablation in the 3D organotypic model was further confirmed in the tumor-bearing mouse models [66].
If we are able to mimic the active TME on multicellular OOCs, can we use these platforms to model PDAC drug response in vitro? A humanized microfluidic device, where PSCs were cultured with PANC-1 cells, represented expected histologic features of PDAC and showed the potential adjuvant therapeutic activity of anti-TME agents to conventional chemotherapies [
115]. The data suggest that besides studying cytotoxicity, this model has the potential to determine the effects of TME compactness and collagen reorganization on PDAC therapeutics [
115]. A similar drug response to Cisplatin has been shown in a microfluidic chamber cultured with different PDAC cells in ECM-enriched environments [
116].
Moving towards a patient-based ex vivo preclinical platform, work is in progress to use tissue-driven cells in OCC models of cancer. In this regard, organoids have made it possible to test drugs on the individual’s tumor cells in the lab. Next, we will discuss how the integration of organotypic technologies in OOCs could allow us to model complex cellular interactions, and the therapeutic activity of anticancer drugs, with the potential to design novel therapeutics at the individual level.
3. Individualized PDAC Model on Chip
The variation in sensitivity to anticancer drugs among different patients highlights the requirement for more precise treatment selection [
117]. We and others have shown that organoids retain a high degree of similarity to the original tissue, including PDAC [
9,
10]. PDOs are proposed to provide an opportunity for a personalized in vitro platform to test drug sensitivity in individual patients [
118]. In PDAC, organoid technology is instrumental in optimizing the use of sparse tissue collected from clinically indicated endoscopic fine needle biopsies (FNBs) performed for tissue diagnosis. This circumvents the particular challenges to precision medicine in PDAC, which stems from limited access to surgically-naïve specimens for pre-treatment screening (>80% of PDACs are unresectable) and rapid patient deterioration [
119]. While organoids are superior to conventional cells for predicting drug response, they often show uncertain growth and considerable heterogeneity and are challenging to manipulate using conventional in vitro techniques [
117]. Therefore, culturing them in microfluidic OOC platforms that mimic 3D tissue architecture and better facilitate nutrient and gas exchange could be more faithful in modeling the disease [
120]. Work is in progress to make the organoid-based models more complex to simulate in vivo tissue structures [
2,
121].
Such a personalized in vitro chip model was recently developed using PDOs derived from PDAC tumor biopsy, fibroblasts, and endothelial cells tri-cultured in a perfusable 96-well based OOC system [
25]. Symbiotic interaction between the PDOs and fibroblasts was observed with an elevated proliferation and increased PDO diameter in the co-culture system. Moreover, fibroblast contributed to chemoresistance to gemcitabine by secreting collagen, which added to the matrix stiffness and acted as a physical barrier to drug delivery. This co-culture platform showed the importance of the relationship between patient cells and desmoplastic ECM and provided a better understanding of chemotherapeutic agents’ bioavailability inside vascularized tumor tissues. Such a model could demonstrate a particular anticancer drug’s sensitivity to an individual patient in the lab before applying in the clinic.