Oleanolic acid (3β-hydroxyolean-12-en-28-oic acid) is a pentacyclic triterpenoid, widely occurring in the plant kingdom, which includes edible and medicinal plants. The richest source of oleanolic acid (OA) are the leaves of the olive plant. Common culinary spices such as garden thyme and clove plants, as well as fruits, are also sources of OA.
- oleanolic acid derivatives
- Nrf2
- NF-κB
- NSAIDs
- inflammation
- oleanolic acid derivatives conjugates
1. Introduction
Moreover, naturally occurring OA serves as a scaffold for additional modifications to achieve synthetic pentacyclic OA triterpenoids. Over the past years, a large number of triterpenoids have been synthetically modified in order to increase their bioactivity and their protective or therapeutic effects. Moreover, attempts to conjugate synthetic triterpenoids with non-steroidal anti-inflammatory drugs (NSAIDs) or other active components have been made.
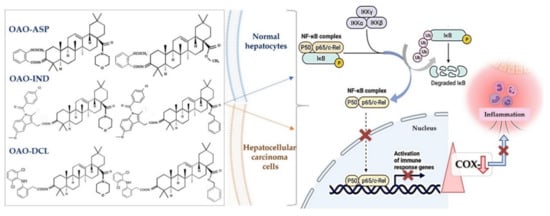
Conjugation with NSAIDs is justified by the fact that hybridization is considered a leading synthetic trend in medicinal chemistry strategy on the one side and the inflammatory background of many types of cancer, such as HCC on the other. NSAIDs are supposed to play a role in cancer treatment and chemoprevention. However, most of these drugs act as COX-2 inhibitors, which, besides their beneficial effects of decreasing the risk of certain types of cancer, are well known for many unfavorable side effects. Fusing NSAIDs with the active oleanane structure, resulting in combined compounds, may enhance the biological effect with synergistic action and minimized toxicity or adverse reactions [4].
The key role in the inflammation process is played by transcription factor NF-κB, controlling the expression of genes such as COX-2 and iNOS , encoding cyclooxygenase-2 and inducible nitric oxide synthetase. Overexpression of COX-2, related to increased cell growth and invasiveness, is observed in human cancers. Downregulation of NF-κB negatively interferes with the Nrf2 signaling pathway, the major cellular defense against reactive oxygen species (ROS) and electrophilic species [5]. Modification of these signaling pathways is usually linked with the other pathways, leading to induction to apoptosis and inhibition of cell proliferation/survival. In recent years, excellent reviews have been published on synthetic triterpenoids, starting with the comprehensive work of the pioneers in this field, Liby and Sporn [6].
2. Tumor Microenvironment and Signaling Pathways—Possible Targets of Oleanolic acid Derivatives
The critical role of the tumor microenvironment in the process of carcinogenesis is now widely accepted. Inflammatory cells, macrophages, neutrophils, or lymphocytes have the ability to generate highly reactive species, which directly damage DNA and can lead to the initiation of carcinogenesis and subsequently promote clonal growth of the initiated cells [7,8]. Moreover, inflammatory cells contribute to the process of angiogenesis [8]. Therefore, chronic inflammation, which promotes angiogenesis and is linked to the development of at least 30% of all cancers, is an important target of cancer prevention and therapy. Consequently, anti-inflammatory drugs are currently repurposing into oncological applications [7,9].
After translocation into the nucleus, the NF-κB complex binds to a specific DNA target sequence (5′-GGGRNYYYCC-3′) [15], assembles with the basal transcriptional machinery, and might associate with other transcription factors, including AP-1 (c-Jun/c-Fos complex) and chromatin remodeling proteins, such as CREB-binding protein (CBP) and p300 [16]. A variety of factors can influence and activate NF-κB nuclear translocation, and thus will be responsible for the diverse and sometimes opposing roles of NF-κB as a pro-and an anti-inflammatory mediator [17]. In this regard, NF-κB is well known for providing cancer cells with a survival advantage by upregulating anti-apoptotic genes. Moreover, there is reciprocal cross-talk between NF-κB and autophagy in cancer. It can either promote or repress tumorigenesis, depending on the stimulus and the context [18].
The persistent activity of NF-κB in tumors is closely associated with the STAT3 transcription factor [19]. STAT3 enhances p300-mediated RelA acetylation, leading to the nuclear retention of NF-κB. Thus, persistently activated STAT3 maintains the constitutive NF-κB activity in tumor cells [20]. STAT3 signaling is a major intrinsic pathway for cancer inflammation due to its frequent activation in malignant cells, with a key role in regulating numerous genes crucial in inflammation-induced cancer and the tumor microenvironment. In this regard, aberrant STAT3 promotes uncontrolled cellular growth and survival through dysregulation of gene expression, including cyclin D1, c-Myc, Bcl-xL, Mcl-1, and thereby contributing to cells surviving. STAT3, besides the mechanism mentioned above, interacts with NF-κB at multiple levels; it is activated by several NF-κB-regulated gene products, e.g., interleukin-6 (IL-6). Thus, these two transcription factors regulate a multitude of genes essential for cancer-promoting inflammation. Similarly, although to a lesser extent, persistent activation of STAT5 in diverse human cancers increases proliferation, survival, angiogenesis, and metastasis [21].
The interconnection of signaling pathways such as Nrf2, NF-κB, and STAT3, and aberrations in their activation mechanisms, make it possible to use them as a target in cancer cells for potential chemotherapeutic and/or chemopreventive compounds. This approach fits in a new trend called anakoinosis [26] , the name of which derives from the ancient Greek term for “communication.” The concept of anakoinosis is an alternative to conventional chemotherapy, which is usually based on a single target, or focused on a single area of the tumor. Unlike standard therapies, treatment protocols based on anakoinosis are less likely to lead to drug resistance.
3. Overview of Oleanolic Acid Derivatives—Chemical Approach
The synthesis of naturally occurring pentacyclic triterpenes with at least eight chiral centers, in particular OA, has been at the center of intense research. Chemical modifications of its structure to obtain synthetic OA derivatives are carried out to increase its potency, reduce toxicity, and increase bioavailability and solubility. Chemical modifications have led to a series of derivatives, such as 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid (CDDO) which, in certain aspects, is over 200,000 times more potent than the parent OA [27,28]. Other promising structures are different esters and oximes [29,30].
OA is characterized by two methyl groups at C-4, one at C-8, one at C-10, one at C-14, and two at C-20. In addition, a carboxyl group is located at the C-17 position, and a double bond is present between C-12 and C-13. Moreover, the stereochemistry of the -OH group at the C-3 position has important physiological implications. In this regard, the less common 3α-OH isomers have different biological activities not shared by the more common 3β-OH isomers. These steric properties of the exocyclic methyl and other groups on natural triterpenoids are essential determinants of their activity and safety in pharmacological contexts and, therefore, became targets of modification [6].
The pioneering work of the Sporn and Liby group [6,31,32,33] and that which followed provided hundreds of synthetic OA derivatives, some of which were patented. Chemical modifications of OA at the C-3 hydroxyl group, at the C-12 to C-13 double bond, and at the C-28 carboxyl group led to new synthetic compounds that are many thousand-fold more active than the parent oleanolic acid, with useful pharmacokinetic and pharmacodynamic profiles. Modifications of the A and C rings to enones, of the C-2 substitution, of C-28 with a wide variety of esters and amides, and the modifications of the six-membered carbon ring A and E represent the most common modifications, leading to promising synthetic oleanolic acid derivatives ( Figure 1 ) [30].
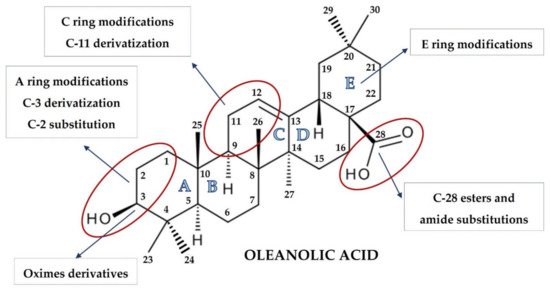
The most extensively explored OA derivatives are CDDO and its derivatives modified at the C-17 position: methyl ester (CDDO-Me; bardoxolone methyl), imidazolide (CDDO-Im), di-CDDO (nitrile at the C-17 position of CDDO; TP-225), and various amides (methyl amide, CDDO-MA; ethyl amide, CDDO-EA; and trifluoroethyl amide, CDDO-TFEA) [6,30]. The results of several studies have pointed out that the potential therapeutic activities of OA derivatives were correlated with their hydrophilicity. Although OA bears a hydroxyl group at the C-3 position and a carboxyl group at the C-17 position, the contribution of these two hydrophilic groups to the overall hydrophilicity of OA is limited. It might be assumed that hydrophilic moieties, coupled with certain long, flexible spacers, could improve OA′s hydrophilicity [34]. Therefore, a series of OA oximes were synthesized with different substituents at the C-17 position: basic carboxyl group, methyl ester, benzyl ester, or morpholide group, assuming this would provide higher hydrophilicity and bioavailability [35].
4. Conjugation of Synthetic OA Derivatives May Enhance Their Anti-Cancer Potential
Considering the inflammatory backgrounds of several cancers, non-steroidal anti-inflammatory drugs (NSAIDs) are supposed to be useful in cancer treatment. Numerous reports concerning the cancer-protective effects of NSAIDs have been published. Now, emerging evidence indicates that such drugs may have activity not only in chemoprevention, but also in treatment [9]. However, most of these drugs act as COX-2 inhibitors which, besides beneficial activity, decreasing the risk of certain types of cancer, are well known for many unfavorable side effects. In addition, their long-term use is often associated with many serious cardiovascular, gastrointestinal, renal, and other side effects [59].
This effect in HepG2 cells was less pronounced in comparison with non-conjugated OAO. In contrast, conjugation enhanced Nrf2 activation in normal hepatocytes of the THLE-2 cells line. These results indicated that OAO derivatives conjugated with ASP have the potential for application in liver cancer chemoprevention. OAOs themselves, particularly OAO substituted with morpholide, may be considered therapeutic agents, which may support conventional treatment strategy [55]. In contrast to OAO conjugates with ASP, OAO hybrids with indomethacin and diclofenac reduced activation and expression, not only NF-κB , but also Nrf2 in hepatoma cells, while in normal cells, increased activation of Nrf2 was still observed ( Figure 3 and Figure 4 ) [39,62].
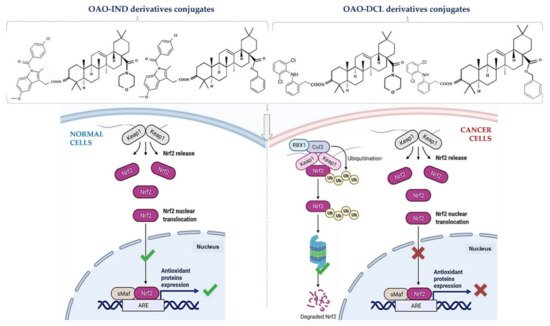
NF-κB negatively interferes with the Nrf2 signaling pathway, and often agents that suppress NF-κB signaling activate Nrf2 [63]. Although the Nrf2-ARE pathway plays a cytoprotective role in normal cells, Nrf2 is frequently overexpressed in cancer cells, including HCC, and may contribute to increased proliferation, invasion, and chemo radio-resistance [64,65,66]. Therefore, the reduced activation of Nrf2 and increased ROS production resulting from treatment with OAO conjugates with indomethacin or diclofenac may protect cancer cells against chemoresistance inhibition of the Nrf2-ARE pathway and, at the same time, exert a chemopreventive effect in normal hepatocytes.
Moreover, it was found that CDDO-Me protected human colon epithelial cells against radiation-induced DNA damage through activation of Nrf2. Therefore, it may protect the gastrointestinal tract against acute irradiation [50]. Thus, OAO conjugates with NSAIDs may increase therapeutic efficacy in cancer cells while protecting normal cells in their microenvironment. The final effect depends on the type and structure of the particular class of NSAIDs.
This entry is adapted from the peer-reviewed paper 10.3390/molecules26164957