Complete androgen insensitivity syndrome (CAIS) is due to complete resistance to the action of androgens, determining a female phenotype in persons with a 46,XY karyotype and functioning testes.
- complete androgen insensitivity syndrome
- androgen receptor
- AR gene
- gonadal neoplasia
- gonadal removal
1. Introduction
Androgen insensitivity syndrome (AIS; OMIM#300068; ORPHA99429; ICD10-E34.5) is a main disorder (or difference) of sex development (DSD) with a 46,XY karyotype[1][2][3]. Albeit rare (with an estimated prevalence of 1:20,000–1:100,000 births)[1][3][2][4], AIS likely represents the most frequent 46,XY DSD, ranging from 40 to 80% in some series[4][5][6][7]. AIS is characterized by the presence of male gonads (testes) in subjects with an external female phenotype but absent internal genitalia (complete AIS or CAIS) or with varying degrees of undervirilization of the internal and/or external genitalia[1][2][3].
2. Molecular Biology
The androgen receptor (AR) protein belongs to the superfamily of nuclear receptors also designated as NR3C4 (nuclear receptor subfamily 3, group C, member 4). The AR protein consists of a 920 amino acid sequence, has a molecular mass of 110 kDa and is organized in eight exons (indicated as A–H or 1–8) and seven introns. The AR receptor is a single-stranded polypeptide consisting of four main structural domains[1][2][3][8][9]. The N-terminal domain (NTD, 538 amino acids) is encoded by exon 1 and contains the activation function-1 (AF-1) region. It is the transactivating domain, which starts and regulates the transcription of target genes and contributes to the final three-dimensional structure of the receptor[2]. The DNA-binding domain (DBD, amino acids 558–617), encoded by exons 2 and 3, is made up of a high number of cysteine residues which bind two zinc atoms through disulfide bridges, resulting in a tertiary structure called the “zinc finger”, particularly suitable for binding with hormone response elements (HREs). The hinge domain, containing the phosphorylation site for AR, is responsible for androgen-dependent structural changes. The ligand-binding domain (LBD, amino acids 646–920), encoded by exons 4–8, contains specific binding sites for androgens, various transcription factors of coactivation and the activation function-2 (AF-2) region. It promotes the interaction of the receptor with the heat shock proteins (HSPs) in the cytoplasm and then with the androgen hormone, leading to the migration of the AR into the nucleus[2][8][9][10].
A unique feature of the AR receptor is the N-terminal–C-terminal interaction between the AF-1 (N-terminal) and AF-2 (C-terminal) subdomains, aimed to stabilize the connection between the receptor and its ligand and to slow down its dissociation. AF-1 acts in a ligand-independent manner, while AF-2 is ligand-dependent and binds to p160 steroid receptor coactivators such as SRC1, SRC2/TIF2 and SRC3. The homopolymeric traits of amino acids within the NTD (CAG and GGN) are independent modulators of the receptor activity. The three-dimensional structure of the receptor comprises 12 α-helices associated with folded β-sheets; they are arranged as a “tripartite sandwich”. The hydrophobic binding pocket is formed by the helices 3, 4, 5, 7, 11 and 12. Helix 12 (H12) is the outermost α-helix, which folds over the top of the hydrophobic pocket like a box lid. This allows the receptor to “capture” the ligand and hold it, slowing the rate of dissociation, according to an effect called the “mouse trap”. It allows the interaction between the LBD domain, the AF2 subdomain and the LXXLL motif of the associated coregulatory proteins [2][8][9][10].
In baseline conditions, AR resides in the cytoplasm where it forms a multimeric complex with heat shock proteins (HSPs), especially with HSP70, HSP90 and HSP56. After binding with androgens, the receptor dissociates from these proteins, dimerizes and translocates into the nucleus. Nuclear transport is selective and active; it consists of two steps, of which the first does not require energy, while the second step depends on the presence of adenosine triphosphate (ATP). Once in the nucleus, the androgen–AR complex interacts with HREs. The interaction with specific (e.g., ARA24, ARA54, ARA55 and ARA70) and nonspecific (e.g., the SRC and CBP/p300 family of proteins) coactivators and corepressors is fundamental in this complex network[2][3][9][8][10].
According to some AR mutation databases, hundreds of pathogenetic mutations related to CAIS are known (Table 1) [11]. Complex rearrangement has been rarely reported.
Table 1. AR genetic variants in complete androgen insensitivity syndrome (CAIS): Pisa Pediatric Endocrinology experience in comparison with international databases[11].
| Type of Genetic Variants | Pisa a | McGill b | HGMD c |
|---|---|---|---|
| Point missense or nonsense mutations | 65.1% | 50.0% | 75.0% |
| Insertions or deletions | 12.1% | 28.0% | 6.6% |
| Intronic or intron–exon junction point mutations | 12.15% | 5.4% | 4.0% |
| Small or gross deletions of the AR gene | 7.6% | 6.7% | 14.4% |
| Complete deletions of the AR gene | 3.0% | 1.3% | ¾ |
Genetic variants associated only with CAIS (no partial or minimal AIS considered): a n = 66; Pisa Pediatric Endocrinology series partly published in 4; b McGill University AR database: n = 314, (www.mcgill.ca/androgendb); c HGMD (Human Gene Mutation Database): n = 533 (www.hmgd.cf.ac.uk).
The majority of AR mutations (about two-thirds) are of germline origin inherited from asymptomatic mothers; in other cases, CAIS is due to somatic and de novo mutations[12]. It has been estimated that up to one-third of the de novo mutations may arise in the postzygotic phase, which could partly explain the phenotypic variability observed in subjects with the same genetic defects. In the absence of any mutation in the AR gene, but in the presence of the phenotype as well as biochemical data suggesting AIS, an altered signaling pathway due to the impairment of some coactivators or some postligand binding factor has been suggested. However, mutations in cofactor genes have not been found in the large majority of such cases; this hypothesis will require more investigations[13].
3. Clinical Features
Females with CAIS present with a normal external female phenotype in girls and women with a 46,XY karyotype and normally functioning testes[2][1][3][8][9]. Psychosexual development is in agreement with female sex[8]. The internal genitalia are absent (“empty pelvis syndrome”) due to the normal action of the anti-Müllerian hormone (AMH) produced by Sertoli cells before birth, which causes regression of the Müllerian structures (uterus, cervix and proximal vagina). Moreover, Wolffian structures do not differentiate because of testosterone resistance[2][1][3]. The testes can be in the abdomen, in the inguinal canal or in the labia majora, causing a bilateral inguinal hernia or labial swelling. These findings are the most frequent clinical signs to suspect CAIS in prepubertal girls. Audi et al. [14] reported inguinal hernia in 47.8% of their sample as a cause for medical advice. Nearly 57% of the CAIS population presented with an inguinal hernia in the U.K. series[15]. In Pisa series, an inguinal hernia was the cause for referral in more than 30% of cases (17/53). The incidence of inguinal hernias in the pediatric population is 1–4% with a clear prevalence in males (10:1). Thus, karyotypes should be performed in girls with a mono- or bilateral inguinal hernia [15]. At puberty, there is normal breast development and a typical female distribution of adipose tissue due to androgens being aromatized to estrogens. However, pubic and axillary hair is usually absent or may be scanty. The vagina has a blind bottom, with a length ranging from 2.5 to 8 cm; it is usually adequate for sexual intercourse. Primary amenorrhea, owing to the absence of a uterus, represents the second main reason for medical consultation [1][2][3][8][9].
The diagnosis of CAIS can also arise because of a mismatch between the prenatal sex (based on free-fetal DNA and karyotype analysis) and the phenotype at fetal ultrasound scans or at birth. Some series reported this clinical presentation in about 3%[14] to 5.6%[4]. An increase of prenatal diagnosis will occurr because the use of prenatal screening tests is spreading. The need for experienced medical staff to counsel the parents on this delicate area will be urgent. Diagnosis may also be the consequence of a known family history of CAIS (4.3% according to Audi et al. [14]and 13% in Pisa experience[4]).
In consideration of the X-linked transmission, it is recommended to perform a karyotype in prepubertal female relatives of a proband, given the possible recurrence of CAIS within the same family (Figure 1).
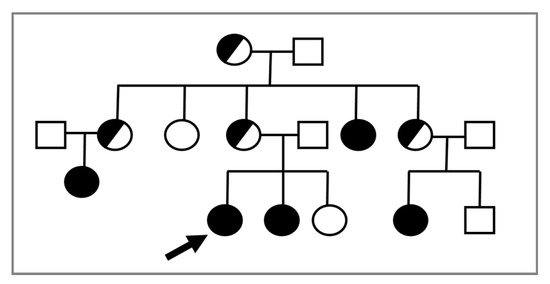
Figure 1. The proband (indicated by the arrow) is a young 46,XY woman (25 years old) with CAIS, in whom the AR variant p.Trp797* was found. Subsequently, the same mutation was detected in the 46,XY sister with CAIS (black circle) and in other affected 46,XY family members with the same DSD (black circles). Female 46,XX carriers (half black circles) were suspected but not proven by molecular analysis of AR.
A delay between the initial clinical suspicion and the definitive diagnosis is still present despite advances in molecular techniques in some experiences[4][6]. Delayed diagnosis and surgery are performed without seeking a definitive diagnosis in other series[7]. About 10% of misdiagnoses was recently reported in a group of women with a previous “tag” of CAIS, suggesting that re-evaluation of old diagnoses will enable a better definition of the clinical picture of 46,XY DSD[4].
4. Oncological Risk
Malignant transformation of the gonads is the most feared complication in women with CAIS[3][8][9]; timing of gonadectomy to prevent cancer is an issue of debate[16].
Gonadectomy has been performed prior to definitive molecular diagnosis in some females, and early gonadal removal may still occur in prepubertal girls[4][17][18]. However, the oncological risk in children with CAIS is relatively low and remains low until the age of majority (0.02–3%). Deans et al.[19] found that the neoplastic risk is around 0.02% in women under 30 years old and up to 22% in those over that age. Chaurdy et al.[17] reported a neoplastic risk between 0.8 and 22%, with an overall risk of approximately 1.5% in 133 patients over 20 years of age[17]. Thus, gonadal surgery could be likely delayed until complete pubertal development, permitting a spontaneous growth spurt, spontaneous puberty and autonomous decision about surgery after the achievement of the age of majority[18][20]. At any rate, gonadectomy after puberty is still discussed controversially[20].
Postponing gonadectomy until after the age of majority requires an accurate follow-up. Döhnert et al.[20]proposed a regular (bi)annual screening program comprising gonadal imaging by ultrasound or magnetic resonance and the determination of some tumor markers (α-fetoprotein, βHCG, LDH and optionally PLAP in nonsmokers) as well as endocrine evaluation (LH, FSH, testosterone and inhibin B). None of these serological markers are able to detect early neoplastic degeneration of the gonads[21], but the development of specific microRNA assays will be an accurate and sensitive method for the early recognition of a gonadal tumor[21][22]. A major candidate is miR-371a-3p; it is relatively close to being introduced in clinical practice for malignant giant cell tumors (GCTs)[22]. Voorhoeve et al.[23] demonstrated that the members of miR-371-3 can be alternative inhibitors of the p53 pathway. MiR-371a-3p is highly informative in identifying the malignant component of all GCTs except teratoma compared to the standard α-fetoprotein and βHCG. It can be detected in serum, plasma and cerebrospinal fluids[23]. MiR-375 has been suggested to be diagnostic for teratoma as well, although it is not clinically proven so far[24].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22031264
References
- Quigley, C.A.; De Bellis, A.; Marschke, K.B.; el-Awady, M.K.; Wilson, E.M.; French, F.S. Androgen receptor defects: Historical, clinical, and molecular perspectives. Endocr. Rev. 1995, 16, 271–321.
- Galani, A.; Kitsiou-Tzeli, S.; Sofokleous, C.; Kanavakis, E.; Kalpini-Mavrou, A. Androgen insensitivity syndrome: Clinical features and molecular defects. Hormones 2008, 7, 217–229.
- Hughes, I.A.; Werner, R.; Bunch, T.; Hiort, O. Androgen insensitivity syndrome. Semin Reprod. Med. 2012, 30, 432–442.
- Costagliola, G.; Cosci O di Coscio, M.; Masini, B.; Baldinotti, F.; Caligo, M.A.; Tyutyusheva, N.; Sessa, M.R.; Peroni, D.; Bertelloni, S. Disorders of sexual development with XY karyotype and female phenotype: Clinical findings and genetic background in a cohort from a single centre. J. Endocrinol. Investig. 2021, 44, 145–151.
- De Paula, G.B.; Barros, B.A.; Carpini, S.; Tincani, B.J.; Mazzola, T.N.; Sanches Guaragna, M.; Piveta, C.S.; de Oliveira, L.C.; Andrade, J.G.; Guaragna-Filho, G.; et al. 408 cases of genital ambiguity followed by single multidisciplinary team during 23 years: Etiologic diagnosis and sex of rearing. Int. J. Endocrinol. 2016, 2016, 4963574.
- Walia, R.; Singla, M.; Vaiphei, K.; Kumar, S.; Bhansali, A. Disorders of sex development: A study of 194 cases. Endocr. Connect. 2018, 7, 364–371.
- Berglund, A.; Johannsen, T.H.; Stochholm, K.; Viuff, M.H.; Fedder, J.; Main, K.M.; Gravholt, C.H. Incidence, prevalence, diagnostic delay, and clinical presentation of female 46,XY disorders of sex development. J. Clin. Endocrinol. Metab. 2016, 101, 4532–4540.
- Batista, R.L.; Costa, E.M.F.; Rodrigues, A.S.; Gomes, N.L.; Faria, J.A., Jr.; Nishi, M.Y.; Arnhold, I.J.P.; Domenice, S.; Mendonca, B.B. Androgen insensitivity syndrome: A review. Arch. Endocrinol. Metab. 2018, 62, 227–235.
- Gulía, C.; Baldassarra, S.; Zangari, A.; Briganti, V.; Gigli, S.; Gaffi, M.; Signore, F.; Vallone, C.; Nucciotti, R.; Costantini, F.M.; et al. Androgen insensitivity syndrome. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3873–3887.
- Gobinet, J.; Poujol, N.; Sultan, C. Molecular action of androgens. Mol. Cell. Endocrinol. 2002, 198, 15–24.
- Gottlieb, B.; Beitel, L.K.; Nadarajah, A.; Paliouras, M.; Trifiro, M. The androgen receptor gene mutations database: 2012 update. Hum. Mutat. 2012, 33, 887–894.
- Hiort, O.; Sinnecker, G.H.; Holterhus, P.M.; Nitsche, E.M.; Kruse, K. Inherited and de novo androgen receptor gene mutations: Investigation of single-case families. J. Pediatr. 1998, 132, 939–943.
- Achermann, J.C.; Hughes, I.A. Disorders of sexual development. In William Textbook of Endocrinology, 11th ed.; Kronenberg, H.M., Melmed, S., Polonsky, K.S., Larsen, P.R., Eds.; WB Saunders: Philadelphia, PA, USA, 2008; pp. 800–836.
- Audi, L.; Fernández-Cancio, M.; Carrascosa, A.; Andaluz, P.; Torán, N.; Piró, C.; Vilaró, E.; Vicens-Calvet, E.; Gussinyé, M.; Albisu, M.A.; et al. Novel (60%) and recurrent (40%) androgen receptor gene mutations in a series of 59 patients with a 46,XY disorder of sex development. J. Clin. Endocrinol. Metab. 2010, 95, 1876–1888.
- Deeb, A.; Hughes, I.A. Inguinal hernia in female infants: A cue to check the sex chromosomes? BJU Int. 2005, 96, 401–403.
- Bertelloni, S. Gonadal surgery in complete androgen insensitivity syndrome: A debate. Sex Dev. 2017, 11, 169–170.
- Chaudhry, S.; Tadokoro-Cuccaro, R.; Hannema, S.E.; Acerini, C.L.; Hughes, I.A. Frequency of gonadal tumors in complete androgen insensitivity syndrome (CAIS): A retrospective case-series analysis. J. Pediatr. Urol. 2017, 13, 498.e1–498.e6.
- Tack, L.J.W.; Maris, E.; Looijenga, L.H.J.; Hannema, S.E.; Audi, L.; Köhler, B.; Holterhus, P.M.; Riedl, S.; Wisniewski, A.; Flück, C.E.; et al. Management of gonads in adults with androgen insensitivity: An international survey. Horm. Res. Paediatr. 2018, 90, 236–246.
- Deans, R.; Creighton, S.M.; Liao, L.M.; Conway, G.S. Timing of gonadectomy in adult women with complete androgen insensitivity syndrome (CAIS): Patient preferences and clinical evidence. Clin. Endocrinol. 2012, 76, 894–898.
- Döhnert, U.; Wünsch, L.; Hiort, O. Gonadectomy in complete androgen insensitivity syndrome: Why and when? Sex Dev. 2017, 11, 171–174.
- Cools, M.; Looijenga, L. Update on the pathophysiology and risk factors for the development of malignant testicular germ cell tumors in complete androgen insensitivity syndrome. Sex Dev. 2017, 11, 175–181.
- Looijenga, L.H.J.; Kao, C.S.; Idrees, M.T. Predicting gonadal germ cell cancer in people with disorders of sex development; insights from developmental biology. Int. J. Mol. Sci. 2019, 20, 5017.
- Voorhoeve, P.M.; le Sage, C.; Schrier, M.; Gillis, A.J.; Stoop, H.; Nagel, R.; Liu, Y.P.; Van Duijse, J.; Drost, J.; Griekspoor, A.; et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell 2006, 124, 1169–1181.
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.J.; Newton, Y.; Hegde, A.M.; et al. Integrated molecular characterization of testicular germ cell tumors. Cell Rep. 2018, 23, 3392–3406.