2. Basic Principles of SUMOylation and Its Role in Physiology
Small ubiquitin-like modifiers (SUMOs) are a group of ubiquitin-like small proteins that are attached to substrate proteins in a reversible post-translational modification termed SUMOylation that is highly conserved in eukaryotes [
9,
10]. The SUMO pathway is involved in several central cellular processes. Modification by SUMO mediates the protein–protein interactions of target substrates, and influences their subcellular localization, stability, and enzymatic function [
9,
11]. SUMOylation can simultaneously target several members of a protein group to modulate the activity of a specific pathway [
12]. The SUMO pathway shares some structural and functional similarities with the ubiquitin pathway [
11]. The global molecular structure of SUMO is similar to ubiquitin, and both have a ββαββαβ fold structure and a conserved position of a diglycine (GG) motif required for isopeptide bond formation [
13,
14].
To date, five isoforms of SUMO (SUMO1, 2, 3, 4, and 5) have been identified in the human genome [
9]. SUMO1, SUMO2, and SUMO3 are ubiquitously expressed in tissues, while the expression of SUMO4 and SUMO5 is restricted only to specific tissues [
9,
15,
16,
17,
18]. SUMO1, SUMO2, and SUMO3 account for most of SUMO modifications, whereas the functional roles of SUMO4 and SUMO5 are more unclear [
15,
18]. SUMO2 and SUMO3 both share 97% sequence identity, but share only 50% sequence similarity with SUMO1 [
19]. Expression levels of SUMOs are dynamic and fluctuate between different developmental stages [
17]. SUMOs can be covalently attached to a single lysine residue (monoSUMOylation) or to multiple lysine residues (multiSUMOylation) of the substrate (
Figure 1). SUMO is preferentially attached to SUMO consensus ΨKxE motifs of substrates, in which Ψ denotes a hydrophobic amino acid, K is lysine, x is any amino acid and E is glutamic acid, although some substrates are also SUMOylated at lysines in non-consensus sites [
10,
20,
21]. SUMO itself may also be SUMOylated (polySUMOylation) to form polymeric SUMO chains, which are restricted to SUMO2 and SUMO3 [
22]. SUMO1 does not form chains efficiently and is predominantly associated with monoSUMOylation [
22]. However, SUMO1 can be attached to SUMO2/3 chains, resulting in the termination of chain elongation [
23]. Non-covalent attachment of SUMO is regulated through the SUMO interaction motif (SIM) of binding partners [
24,
25].
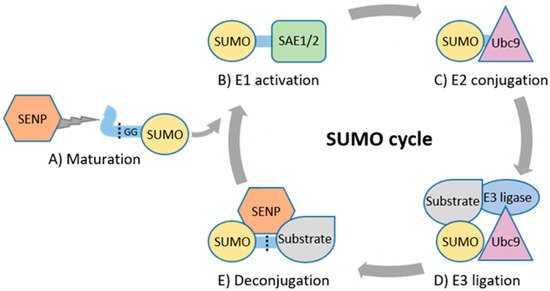
Figure 1. The enzymatic SUMO cascade. (A) SENPs cleave off amino acids from precursor SUMO to produce a mature SUMO. (B) SAE1/SAE2 forms of a thioester bond between SUMO’s GG and catalytic cysteine residue of SAE1/2. (C) Thioester bond is formed between SUMO’s GG and catalytic cysteine residue of Ubc9. (D) Ubc9 catalyzes formation of an isopeptide bond between GG and target substrate’s lysine residue often with assistance of an E3 ligase. (E) SENPs deconjugate SUMO from the substrate.
The enzymatic cascade regulating SUMOylation comprises SUMO E1-activating, E2-conjugating, E3 ligase and deconjugating enzymes (
Figure 1) [
9]. Maturation and deconjugation (deSUMOylation) of SUMO is regulated by the family of cysteine proteases, sentrin-specific proteases (SENP1, 2, 3, 5, 6, 7, and 8) [
26]. SENP1–7 have specific, varying subcellular localizations and regulate SUMO processing, whereas SENP8 displays substrate specificity only for ubiquitin-like protein NEDD8 [
26,
27,
28]. SUMO E1-activating enzyme (SAE1/2) activates mature SUMO and transfers it to the only known mammalian E2-conjugating enzyme, Ubc9 (encoded by
UBE2I) [
29,
30,
31]. Ubc9 alone is capable of conjugating SUMO to substrates in vitro. However, E3 ligase enzymes are required for specificity and efficiency of SUMOylation. The family of protein inhibitors of activated STAT (PIAS) is the major class of SUMO E3 ligases, and the human genome has four PIAS genes (PIAS1, 2, 3, and 4) that encode seven protein products, PIAS1, PIAS2a (PIASxα), PIAS2b (PIASxβ), PIAS3, PIAS3L, PIAS4 (PIASy), and PIASyE6− [
32,
33]. Other classes of SUMO E3 ligases include the well-characterized Ran-binding protein 2 (RanBP2), and few other E3 ligases, such as ZNF451, polycomb protein PC2 (also known as CBX4), and some members of the tripartite motif (TRIM) protein family [
34,
35,
36,
37,
38].
The SUMO pathway undergoes constant cross talk with other post-translational modifications, including phosphorylation and methylation [
39,
40,
41]. For instance, phosphorylation can influence SUMOylation of substrates containing the conserved phosphorylation-dependent SUMOylation motif (PDSM), which mediates phosphorylation-dependent SUMOylation of various transcriptional regulators [
39]. In addition, lysine methylation of the KKxE motif can create a motif resembling the SUMO consensus site in substrates containing the KKxE motif [
40]. HMGA2 SUMOylation is dependent on its methylation at K66 and K67 of KKAE motif by methyltransferase SET7/9. The KKxE motif is present in several other proteins, including the polyhomeotic complex 1 (PHC1), which also requires methylation for its complete SUMOylation.
The importance of SUMOylation in physiology is apparent from early development. A functional SUMO pathway is essential for embryogenesis. Loss of Ubc9 leads to severe defects of chromosome condensation and segregation, as well as disruption of nuclear organization, resulting in death at early embryonic development in mice [
42]. SUMO1 knockout mice are viable, as SUMO2/3 can compensate for most functions of SUMO1 [
17,
43]. Interestingly, SUMO3
−/− mice are also viable, but the knockout of SUMO2 leads to severe developmental defects and early death before adulthood, indicating that SUMO2 is essential for the development. Balance of SUMOylation and deSUMOylation is critical for embryonic development, as knockout of SENP1, SENP2, or SENP3 results in embryonic lethality in mice, suggesting non-redundancy and substrate specificity among SENPs [
44,
45,
46]. SENP1
−/− mice have severe fetal anemia and SENP2
−/− mice display defects in trophoblast development, whereas conditional knockout Senp3
+/− mice have impaired cytokine and inflammatory signaling. PIAS1
−/− and PIAS4
−/− knockout mice are produced at a lower frequency than the expected Mendelian ratio due to increased perinatal lethality, and both knockouts display defects in cytokine signaling [
47,
48]. Surviving PIAS1
−/− mice are runts compared to their wild-type counterparts, whereas PIAS4
−/− mice show no obvious phenotype. PIAS2
−/− knockout mice have reduced testis weight and sperm count but remain viable and fertile [
49]. PIAS3
−/− mice are viable and show no overt phenotype but have an impaired visual response of medium wavelength cones [
50].
3. Altered Expression and Prognostic Significance of SUMO Pathway in Cancer
Dysregulated mRNA and protein levels of the SUMO machinery components have been reported in tissue samples of cancer patients with potential prognostic associations (
Table 1). In the majority of the published reports, the expression levels of SUMO pathway components are upregulated in cancer and are associated with a higher histological grade, higher stage, presence of metastases and poor prognosis. Interestingly, genome-wide analyses have identified concurrently increased levels of multiple SUMO machinery components in certain cancer types [
51]. However, high expression has been associated with better prognosis in some cancer types. Downregulated expression levels in cancer tissues have consistently been reported for SENP2 [
52,
53,
54,
55,
56,
57] and PIAS2 [
58,
59,
60]. The prognostic value of a single SUMO machinery component can also vary between cancer (sub)types, and, e.g., the value for PC2 [
61,
62] and PIAS4 [
63,
64] as prognostic biomarkers in breast cancer and for PIAS3 [
65,
66] in mesothelioma remains ambiguous (
Table 1). Bioinformatic analyses of the SUMO machinery protein and mRNA levels, and their association with prognosis, utilizing datasets from various databases are generally well in line with the other published analyses of often smaller sample sizes [
67,
68,
69]. Mass spectrometry-based methods might decrease variability in protein level analyses caused by the use of different antibodies, whereas IHC allows analysis of the subcellular localization of a protein, which might be crucial for the prognostic significance of the SUMO machinery components in particular [
63,
70,
71].
Table 1. Prognostic value of SUMO pathway components.
High Expression Associates with
Poor Prognosis |
High Expression Associates with
Good Prognosis |
| Cancer Type |
Protein(s) |
Cancer Type |
Protein(s) |
| adrenocortical |
PIAS3, PIAS4, SAE1, SAE2, SENP1, SENP3, SUMO1, SUMO2, SUMO4 |
bladder |
Ubc9 |
| breast |
PC2, PIAS3, PIAS4, SAE1, SAE2, SENP5, SENP7L, SUMO1, SUMO2, SUMO3, Ubc9 |
breast |
PC2, PIAS1, PIAS4 |
| colorectal |
SAE2, SENP1, SUMO1 |
cervical |
PIAS3 |
| gastric |
PC2, PIAS2, SAE2, SUMO3, Ubc9 |
colorectal |
PC2 |
| glioma |
SAE1, Ubc9 |
gastric |
PIAS1, PIAS4 |
| hepatocellular |
PC2, PIAS2, PIAS3, PIAS4, SAE1, SAE2, SENP1, SENP3, SENP5, SENP6, SUMO2, Ubc9 |
glioma |
PIAS3 |
| leukemia |
SAE1, SUMO3 |
leukemia |
PIAS2, SENP5, SENP7 |
| lung |
PC2, SAE1, SAE2, SENP1, SUMO2/3, SUMO4, Ubc9 |
lung |
PIAS3 |
| melanoma (cutaneous) |
SAE1 |
melanoma (cutaneous) |
PIAS1, SENP5, SENP7 |
| melanoma (uveal) |
SAE1, SAE2, SUMO3 |
melanoma (uveal) |
SENP2, Ubc9 |
| mesothelioma |
PIAS3, PIAS4, SAE1, SAE2, SENP1 |
mesothelioma |
PC2, PIAS3, SENP2 |
| multiple myeloma |
Ubc9 |
ovarian |
PIAS2 |
| osteosarcoma |
PC2, SENP3 |
pancreatic |
SENP3 |
| ovarian |
SENP3, SENP5 |
pheochromocytoma and paraganglioma |
Ubc9 |
| pancreatic |
SENP2, (SUMO1 and SUMO2/3 together), Ubc9 |
renal |
PIAS1, PIAS2 |
| prostate |
PIAS1, SAE1, SENP1, SENP5, SUMO1, SUMO2 |
testicular germ cell |
PIAS2 |
| renal |
PC2, PIAS3, RSUME, SAE1, SENP1, SENP3, SENP5, SUMO1, SUMO2, Ubc9 |
thymoma |
PIAS4, SAE1, SAE2, SENP1, Ubc9 |
| sarcoma |
PC2, PIAS2, PIAS3, SENP6, SENP7 |
|
|
| thyroid |
PIAS2, SAE1 |
|
|
| uterine corpus endometrial |
PC2, SAE2, SENP2, SENP5, SUMO4 |
|
|
Regulation of SUMO Machinery Expression and Activity in Cancer
Dysregulation of SUMO pathway components in cancer tissues is often detectable already at the mRNA level. The expression of SUMO machinery components can be epigenetically altered by DNA methylation. For instance, promoter hypomethylation of SENP6 induces expression of SENP6 in hepatocellular carcinoma (HCC) tissues, and elevated SENP6 mRNA and protein levels are associated with promotion of HCC tumorigenesis [
72,
73]. At the post-transcriptional level, numerous miRNAs are implicated in SUMO regulation, and inverse expression levels of SUMO components and their miRNA-regulators are found in several cancers [
74,
75,
76,
77,
78]. For example, a low expression of miR-145 is correlated with high expression of SENP1 in prostate cancer cells, and introduction of miR-145 causes cell cycle arrest via inhibition of SENP1 [
76]. Oncogenic miRNA-9 and miRNA-181a inhibit PIAS3 in IL-6
high breast cancer to promote expansion of early-stage myeloid-derived suppressor cells, resulting in suppression of T-cell immunity [
79].
Dysregulation of the expression of SUMO machinery components in protein level can also occur in cancer. Post-translational regulation by, e.g., ubiquitin and subsequent proteasomal degradation may be involved, resulting in alterations in expression levels in cancer [
80,
81]. Moreover, reactive oxygen species (ROS) typically present in cancer influence protein expression levels of SENPs [
82,
83]. For instance, the ROS-induced increase in SENP3 level can drive carcinogenesis of head and neck cancer via deSUMOylation and subsequent hyperphosphorylation of STAT3 [
83].
In addition to changes in expression levels, catalytic activity of select SUMO pathway components can directly be regulated by conditions typical in cancer, such as hypoxia [
84,
85]. Hypoxia induces a rapid and reversible inhibition of catalytic activity of SENP1 and SENP3, resulting in altered SUMOylation of a subset of proteins, such as the co-repressor BHLHE40 that is implicated in metabolic reprogramming upon hypoxia.