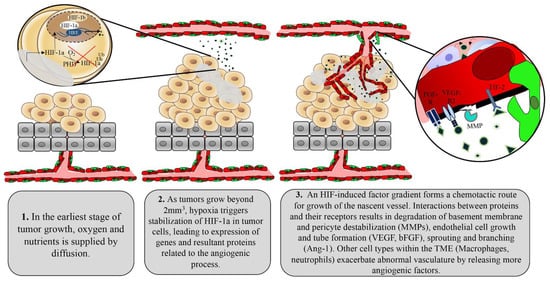
Tumors initiate the angiogenic process through activation of multiple factors including the most prominent angiogenic ligand, vascular endothelial growth factors (VEGF), and its receptors including VEGFR2 [
29,
30,
31]. The VEGF family of proteins includes VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, and placenta growth factor (PIGF) [
31]. VEGF-C and VEGF-D are studied as regulators of lymphangiogenesis, while VEGF-A is commonly referred to simply as VEGF due to its dominant role in angiogenesis. VEGF undergoes alternative splicing, leading to several isoforms that differ based on heparin binding affinity, localization to the extracellular matrix, or diffusive potential. The VEGF gene is transcriptionally regulated in response to HIF, and its levels must be tightly controlled to prevent aberrant angiogenesis [
31]. Due to their control over HIF, tumor cells release exaggerated levels of VEGF to the extracellular space in response to hypoxia [
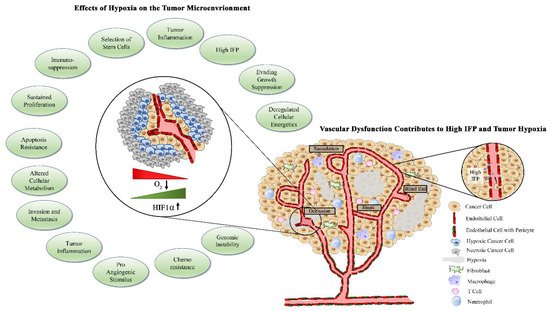
32]. High concentrations of VEGF surrounding endothelial cells select for excess tip cells, which then contribute to irregular branching and tortuous vascular networks. The basement membrane of tumor vessels, which serves as a physical barrier for cancer cell metastasis to surrounding tissues, is often absent or thin due to chemical degradation by tumor-derived proteases [
33,
34]. The monolayer of endothelial cells is often disorganized and cells are plagued with abnormal gene expression profiles, karyotypic abnormalities, and chromosomal instability [
35,
36,
37]. Compared with normal endothelial cells, tumor endothelial cells contain four times the amount of total RNA, indicating enhanced gene expression. Indeed, tumor endothelial cells have enhanced expression of VEGFR-1 and -2 and are therefore more responsive to VEGF stimulation [
38]. Recently, tumor endothelial cells have been shown to have enhanced expression of markers of angiogenesis and stemness such as CD61, CD105, Sca-1, CD34, CD90, and ALDH [
39,
40]. These expression profiles contribute to the escalated angiogenic potential of tumor endothelial cells compared with normal endothelial cells, which facilitates the aberrant vascular arrangement seen in tumors [
41]. Extracellular factors such as VEGF, PMA, TGF-ß, and cytochalasin B, which are overexpressed in the tumor microenvironment, have been shown to impact fenestration formation in endothelial cells [
42,
43]. Given that these plasma membrane microdomains are vital for the exchange of solutes and water at the interface of tissue and vasculature, tumor endothelial cells are often more porous compared with normal counterparts [
43]. Abnormal VEGF signaling in tumor endothelial cells also leads to downregulation of connexin expression, causing gap junction dysfunction, increasing vascular fenestrations, and increasing vascular permeability [
44,
45,
46]. In fact, VEGF was initially identified based on its ability to increase vascular permeability and extravasation of plasma proteins, such as fibrinogen [
47].
Pericytes are specialized smooth muscle cells that are recruited to mature and stabilized vessels through release of PDGF-ß by ECs [
48]. Mice deficient in PDGF-ß signaling lack pericytes and succumb to micro hemorrhaging, demonstrating the importance of these cells for proper vascular function [
48,
49]. Signals secreted by pericytes maintain EC survival by leading to enhanced expression of BCL-w antiapoptotic protein [
49]. Pericytes therefore also shelter normal vessels from anti-angiogenic therapies, allowing for tumor-targeted action of these agents. Hypoxia and downstream angiogenic factors released by tumor cells disengage pericytes from endothelial cells as the initial step to the formation of the nascent vascular sprout. Therefore, tumor-associated vessels are largely devoid of pericytes or demonstrate weak connections between pericytes and endothelial cells, contributing to an immature vascular phenotype and facilitating continued angiogenesis [
50].