One of the most difficult barriers encountered when treating the majority of solid tumours is attributed to the scattered microregions within the tumour characterized by the lack of oxygen. This is known as tumour hypoxia. The lack of oxygen supply results in hypoxic microregions scattered throughout the tumour relative to normal tissue. The hypoxic environments within tumours create several cancer treatment barriers. Most notably, all hypoxic cells are resistant to ionizing radiation (IR). The mechanism by which radiation is able to eradicate tumour cells by damaging DNA, resulting in apoptosis and cell death, occurs through the production of reactive oxygen species (ROS). However, due to the limited oxygen availability within hypoxic tumour microenvironments, this impedes the efficacy of radiotherapy. Furthermore, normal tissues are unable to withstand increased doses of radiation that compensate for tumour hypoxia. Methods targeting tumour hypoxia through radiosensitizing hypoxic cells include hyperbaric oxygen, hypoxic cell radiosensitizers, hypoxic cytotoxins, and tumour metabolism. As research continues to elucidate the relationship between tumour hypoxia and radiotherapy, novel approaches have been developed.
- hypoxia
- non-targeted effects
- autophagy
- PET imaging
- radiosensitizers
1. Tumour Hypoxia Imaging via PET for Guiding Carbogen Breathing Therapy
1.1. Background
One of the earliest techniques used to control tumour hypoxia was hyperbaric oxygen (HBO). Figure 1 shows the rationale behind the modern approach to sensitize hypoxic cells by identifying, imaging, and measuring oxygen levels in hypoxic areas to improve management. The intent of HBO treatment is to increase the supply of oxygen to hypoxic tumour cells. When this treatment is coupled with radiotherapy, this allows the reoxygenated hypoxic tumour cells to become more radiosensitive, thus, reducing the progression of metastasis [1][2][3]. However, due to patient concerns associated with claustrophobia, the time needed for the administration of the treatment, and equal effectiveness using drugs, has led to a shift from hyperbaric oxygen [4][5]. In addition, a systematic review conducted by Bennett et al. [6] suggests more side effects associated with HBO, such as oxygen poisoning and severe radiation injury. Despite the adverse effects associated with HBO, previous studies present conflicting evidence. Particularly, a study conducted by Kohshi et al. [7] suggests that radiotherapy should be performed immediately after HBO treatment, rather than the two procedures occurring simultaneously, to avoid adverse effects from HBO. Based on these conclusions, the use of HBO for treating tumour hypoxia continues to be controversial [8].
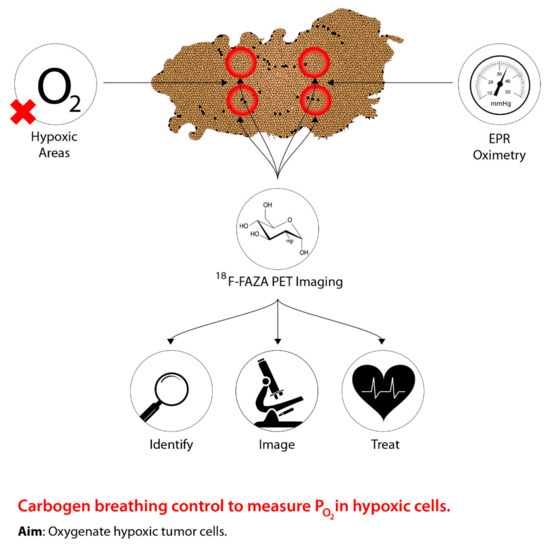
Figure 1. The concept underlying PET imaging to optimize carbogen breathing approaches to defeating hypoxia in radiotherapy. Oxygen levels are measured as the tumour is imaged, thus allowing precise identification of hypoxic regions, which can then be effectively targeted.
The uncertainty surrounding HBO treatment for combating tumour hypoxia has shifted to using a combination of 5% CO2 and 95% O2 to create a gaseous compound known as carbogen [9]. Recent research has shifted away from solely using oxygen manipulation-based methods to guiding carbogen therapy through medical imaging, specifically PET, to predict hypoxic regions of a tumour in order to alter tumour hypoxia therapeutics accordingly [10][11]. Although research has been conducted using various imaging techniques, PET imaging is preferred due to high precision and sensitivity in vivo, as well as providing measurements of intracellular oxygen levels [12]. Moreover, tumour hypoxia imaging using PET allows for identifying novel indicators of tumour hypoxia, as well as aiding in determining baseline responses elicited from hypoxic tumours following hypoxia therapeutics [12]. To facilitate the identification of hypoxic regions within a tumour through PET imaging, a PET radiotracer that is suitable for all types of cancer must be used [12]. However, the most appropriate radiotracer has yet to be identified, but research continues to examine novel and existing compounds in relation to tumour hypoxia imaging and hypoxia therapeutics [12]. Despite this approach being in the early stages, tumour hypoxia imaging can serve as a powerful tool to identify and treat hypoxic tumour microenvironments for cancer therapies relative to normal tissues.
1.2. Mechanism of Action
As previously stated, in order to facilitate effective tumour hypoxia imaging, an “ideal” PET radiotracer is required and must meet a number of biochemical characteristics [12]. Although many compounds are being investigated, virtually all compounds do not meet all the criteria of the “ideal” PET tracer, nor are they available for imaging all tumour types [13]. Despite the lack of an “ideal” PET tracer, research has been focused on nitroimidazole analogs, specifically, 2-nitroimidazole [12]. Although nitroimidazoles were originally intended to be radiosensitizers, Chapman et al. [14] demonstrated that these compounds can serve as hypoxia markers. These compounds are able to passively diffuse into cells, which is largely determined by the intracellular environment [15]. The main driver of the initial reduction following passive diffusion of nitroimidazoles is the concentration of intracellular oxygen [15]. Once the compounds have entered the cell, nitroimidazoles will undergo single-electron reduction to create a free radical anion [15]. Subsequently, within normoxic cells, free radical anions are promptly reoxidized to the parent compound through intracellular oxygen levels due to the high electron affinity relative to the nitro group on nitroimidazole [15]. In contrast, following single-electron reduction of nitroimidazole within hypoxic tumour cells, due to low intracellular oxygen concentrations, reoxidation cannot be completed [15]. Subsequently, incomplete reoxidation results in the further reduction of the free radical anion, creating a very reactive species that are able to bind to components of a cell [15][16]. Furthermore, reduced nitroimidazole has been shown to accumulate within hypoxic cells, thus, demonstrating its potential as a PET tracer [12]. Of the nitroimidazole analogs screened as PET tracers, most compounds are fluorinated nitroimidazoles; however, 18F-fluoromisonidazole (18F-FMISO) has garnered the most success and has been extensively studied [12]. The mechanism of nitroimidazole analogs entrapment within hypoxic tumour cells can be applied to 18F-FMISO. 18F-FMISO is a lipophilic compound; thus, it is readily available to passively diffuse into cells, subsequently, reduction of this compound via the nitroreductase enzyme (NTR), results in the production of R-NO2 radicals [17]. Furthermore, due to the low intracellular oxygen levels (pO2 < 10 mmHg), these radicals are unable to be reoxidized, leading to further reduction of R-NO2 radicals to form R-NHOH molecules that can bind to cellular components, allowing for tumour hypoxia imaging [17][18][19][20]. Recently, another type of fluorinated nitroimidazole, 18F-Fluoroazomycin arabinoside (18F-FAZA), has been gaining more popularity relative to 18F-FMISO [17]. Studies suggest that 18F-FAZA, in comparison to 18F-FMISO, is less lipophilic due to the presence of an additional sugar moiety [17][21][22]. Moreover, due to structural composition differences,18F-FAZA has a faster diffusion and clearance rate relative to 18F-FMISO, allowing for an improved tumour-to-background (T/B) ratio, thus, this compound has been gaining more interest as a PET radiotracer [23][24]. Overall, due to the new spotlight on 18F-FAZA as a PET radiotracer for tumour hypoxia imaging, researchers are beginning to investigate the use of PET imaging to mediate oxygen-based manipulation therapies, such as carbogen breathing. Thus, tumour hypoxia imaging serves as a promising method to identify hypoxic tumour microenvironments relative to normal tissue, ultimately improving the efficacy of oxygen-based manipulation therapies. In relation to the non-targeted effect theme of this review, which is discussed in detail later, it is likely that increased oxygen will increase oxidative stress and lead to initiation of both bystander signalling and genomic instability phenotypes. To what extent this impacts the therapeutic ratio is not known and is an area for further research.
2. Gold Nanoparticles
2.1. Background
Due to the concerns associated with HBO treatment, researchers began to investigate compounds that mimic oxygen, which also allows for the radiosensitization of hypoxic tumour cells. Figure 2 shows the concept behind the modern use of novel hypoxic cell sensitizers. The use of chemical compounds led to the development of a group of compounds classified as nitroimidazoles [25]. Particularly, these groups of drugs are able to differentiate between normal tissue and tumours due to the lack of intracellular oxygen in hypoxic cells [26][27]. Of the four compounds identified, nimorazole was identified as being the least active but displayed the most effectiveness, particularly within patients with head and neck cancers [28][29]. Due to the primitive tumour vasculature system, a diffusion barrier is created between the tumour cells and the blood vessels carrying anticancer drugs, thus, limiting the effectiveness of nitroimidazoles [30][31]. Despite the inviting potential of these drugs, high dosages induce neurotoxic effects on the central nervous system [32]. In addition, Wardman [32] states that some types of nitroimidazoles may reduce the concentration of thiol within normal tissue. Thiols are known to be radioprotective compounds; thus, the depletion of these molecules within hypoxic tumour cells will induce radiosensitization [32]. Consequently, a reduction in thiol concentrations within normal tissue can potentially subject these tissues to radiosensitization, hence, posing detrimental problems [32]. Current research has begun to use nanotechnology to develop novel cancer radiosensitizers comprised of metallic nanomaterials [33]. The integration of nanotechnology within tumour hypoxia therapy is aimed at enhancing the enhanced permeability and retention (EPR) effect, which is a biological dysfunction characteristic of tumours [34]. Due to the novel vasculature system fabricated within tumours, the rapid rate of proliferation leads to the generation of local compressive forces within the vasculature system [35][36]. In particular, the compressive force on the lymphatic vessels results in reduced lymphatic drainage [35]. In addition to the EPR effect, these novel tumour vasculature systems, relative to normal vasculature systems, display larger pores [37]. Due to the large fenestrations, this allows nanomedicines to more easily enter tumours; furthermore, owing to the poor lymphatic drainage systems, nanomedicines that have entered are able to accumulate and take effect within hypoxic tumour regions [37][38][39][40][41]. The primitive characteristics of the tumour-generated vasculature systems are collectively known as the EPR effect and are the target for successful drug delivery [35][42][43]. Wang et al. [33] have indicated that nanomaterials with a high atomic number (Z) are the most promising radiosensitizers due to manufacturing feasibility, size, energy absorption, as well as scattering and emission of radiation energy. In addition, metallic nanomaterials such as gold and silver have demonstrated low toxicity, fast distribution, and agreeable kinetic profiles [33][44][45][46]. Of the metallic nanomaterials, gold nanoparticles (GNPs) demonstrate the most promise due to strong photoelectric absorption, impeccable biocompatibility, and low toxicity [47]. Furthermore, GNPs have a large volume to surface area ratio allowing for other therapeutics to be used, increased effect on EPR, low permeability, contrasting ability in imaging technology, and controlled size distribution [47]. Hence, GNPs themselves and novel drug delivery systems demonstrate promising capabilities as novel radiosensitizers in order to improve targeting to hypoxic tumour sites relative to normal tissue and to decrease toxicity.
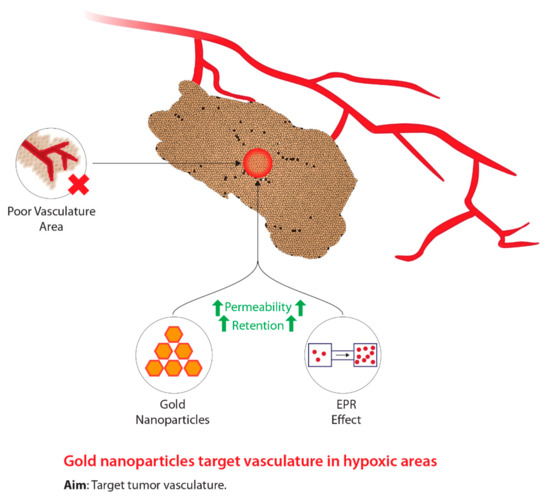
Figure 2. The concept underlying the use of gold nanoparticles in the treatment of tumour hypoxia during radiotherapy.
2.2. Mechanism of Action
Aside from the classic compounds used as radiosensitizers, GNPs have demonstrated promising results as novel hypoxic cell radiosensitizers. Following exposure to IR within hypoxic cells subjected to GNPs, these materials undergo three distinct interactions with IR, physical, chemical, and biological, to induce radiosensitization [44]. First, within nanoseconds of IR exposure, interactions on a physical level begin radiosensitizing hypoxic tumour cells [44]. Due to the discrepancy in energy absorption abilities between gold and soft tissues, gold is an attractive material that can be used to induce physical dose enhancement [44]. Primarily, there are two main mechanisms whereby photons lose energy, namely, the Compton effect and the photoelectric effect [47]. The Compton effect is characterized by the scattering of incident photons caused by colliding with electrons that are weakly held [47]. In addition, during the collision between the incident photon and weakly bounded electrons, the energetic photons will transfer some energy to the electrons, causing the ejection of electrons from the atom [47]. Furthermore, Chen et al. [47] state that in events where the Compton effect is dominant, despite the small amounts of energy transfer, photons retain the majority of the energy and decelerate over long ranges, thus, exhibiting sparse areas of ionization. Contrastingly, the photoelectric effect is distinguishable from the Compton effect because of the strong dependency between the photon energy and electron binding energy [47]. As a result of this dependency, when an incident photon is absorbed by an electron bound to an atom, this leads to the ejection of an inner bound electron [44]. Furthermore, Her et al. [44] explain that in order to compensate for the ejection of an inner bound electron, electrons situated on the outer-shell fall inwards, simultaneously, causing lower energy photons (fluorescence) and a variety of secondary electrons, known as Auger electrons, to be released. Moreover, in order to radiosensitize hypoxic cells, GNPs take advantage of the atom number discrepancy between the high atomic number of gold (Z = 79) and the low atomic numbers of soft tissues [44]. Overall, Her and colleagues [44] state that the difference in atomic numbers allows GNPs to deliver more energy per unit mass, hence, leading to the increased local deposition of radiation within hypoxic areas of a tumour [44].
Following physical interactions with GNPs, chemical interactions soon take effect. Although the mechanisms underlying these interactions have not been completely elucidated, studies suggest that chemical enhancement occurs through two different pathways [44]. The first pathway suggests that DNA becomes chemically sensitized following IR-induced damage, while the secondary pathway suggests that the active surface of GNPs causes the increased formation of radicals and catalysis, leading to chemical sensitization [44]. Chemical sensitization of DNA occurs through the nuclear localization of GNPs to bind to DNA, which causes chromatin structures to “open”, thus, increasing DNA sensitivity to IR [47]. Moreover, electrons with an ionization threshold of <10 eV, known as low energy electrons (LEEs), and secondary electrons are critical for the radiosensitization process [47][48][49]. Although the interaction between LEEs and GNPs does not produce secondary electrons, Chen et al. [47] argue that significant DNA damage can be inflicted. Furthermore, Chen et al. [47] suggest that LEEs cause transient negative ions to weaken the hydrogen bonds within DNA, thus increasing chemical sensitivity. However, it is critical to be cognizant of the charge and size of GNPs, since chemical sensitization depends on these characteristics [44][47][49]. The latter mechanism of DNA chemical sensitization is attributed to the activated surfaces of GNPs, which catalyze a variety of chemical reactions [44][47][50][51]. Specifically, attention has been focused on GNPs (<5 nm) with large surface areas, which demonstrate large catalytic activity governing the transfer of electrons from surface-bound donor groups to O2 to produce free radicals [47]. According to Her et al. [44], due to the small size and curved structure of nanoparticles, this destroys the impeccable structure and organization of gold to produce free radicals on GNPs. Alternatively, the catalytic reactions induced by GNPs can lead to the transfer of electrons and increased production of ROS [47]. Moreover, increased levels of ROS induce negative implications on the biological interactions between GNPs and IR, particularly through oxidative stress [47].
The biological interactions between GNPs and IR occur through three different pathways, oxidative stress, disruption of the cell cycle, and inhibition of DNA repair [44]. One of the primary pathway’s radiation can induce cell killing is through the radiolysis of water, which generate free radicals, and allows ROS to interact with other components of the cell [44]. As described by Her et al. [44], ROS, superoxide radicals (O2−), hydroxyl radicals (OH), and hydrogen peroxide (H2O2) interact with cellular components to induce cellular damage through two different mechanisms. Her and colleagues [44] state that the aforementioned molecules can have direct actions with cell components to directly or indirectly induce oxidative stress, which ultimately triggers cell death through necrosis or apoptosis. Thus, the increased production of ROS mediated by GNPs leads to cell damage through increased oxidative stress, which is the primary characteristic of nanoparticles inducing cytotoxicity [44]. Although the underlying cellular mechanisms are not well understood, recent studies suggest that mitochondria also amplify ROS production [47]. Several groups [52][53] suggest that oxidative stress can induce mitochondrial dysfunction, whereby a multitude of biological effects can lead to apoptosis or necrosis. Chen et al. [47] indicate that GNP-driven oxidative stress leading to increased production of ROS and is linked to mitochondrial dysfunction, which can potentially lead to heightened cell death. Despite the favourable observations, further research must be conducted to elucidate and validate the role of mitochondria and ROS production. Secondly, GNPs disrupt the cell cycle. Within mammalian cells, IR is known to halt cells within the G1 or G2 phase [43]. Moreover, the stages of the cell cycle exhibit various levels of radiosensitivity, whereby cells within the late S-Phase display the maximum radioresitivity, while cells in the G2/M phase are most the radiosensitive [54]. Mackey et al. [55] have elucidated that GNPs have the ability to alter cell-cycle distribution, such that there is an increase in cells within the G2/M phase, hence, increasing radiosensitivity. However, conflicting results from several studies [47][56][57][58] indicate that GNPs do not have an influence on cell-cycle distribution. Evidently, due to the conflicting conclusions between GNPs and cell-cycle distribution, this relationship must continue to be investigated. Finally, IR is known to inflict a variety of DNA damages, namely, double-stranded breaks (DSBs), single-stranded breaks (SSBs), DNA-protein crosslinks, and modifications to DNA bases; however, DSBs are the most lethal [47]. The inability to repair DSBs causes a cascade of cellular impairments that ultimately lead to cell death and can occur in a multitude of ways [47]. Using comet assays in tandem with biomarkers sensitive to DNA damage, such as phosphorylated histone variant γ-H2AX and p53-binding protein 1 (53BP1), this can be employed to uncover the effect of GNPs on DNA repair following exposure to IR [47][59][60][61]. Chen et al. [47] state that several dynamic monitoring experiments using γ-H2AX and 53BP foci assay have been able to detect DNA damage, following the employment of GNPs and IR. Although GNPs’ impact on DNA damage serves as a plausible pathway for radiosensitization, earlier studies have presented conflicting results; thus, further investigations must be conducted to validate the connection [47]. Overall, the physical, chemical, and biological interactions of GNPs leading to radiosensitization have not been completely uncovered. Significantly, no research appears to have been performed looking at the possible involvement of NTE. Metals and other inorganic and organic chemicals are known to produce NTE and to increase genomic instability and bystander effects in vitro and in vivo [62][63][64][65][66][67]. It is probably important to consider whether the mechanisms by which GNPs lead to radiosensitization involve the induction of NTE.
3. Macrophage-Mediated Drug Delivery: HAPs
3.1. Background
Aside from radiosensitizing drugs, alternative drugs classified as hypoxia-activated prodrugs (HAPs) were designed to preferentially kill hypoxic tumour cells through the generation of free radicals [68][69]. The modern approach using macrophages as carriers to deliver HAPs is outlined in Figure 3. The most notable HAP is Tirapazamine (TPZ), which is classified as a benzotriazine-di-N-oxide and has shown compelling results both in vitro and in vivo [68]. However, TPZ has not shown any significant results from clinical studies due to physical concerns, such as severe muscle cramping and nausea [70]. In addition, HAPs may potentially enter normoxic cells and elicit negative effects, demonstrating the inefficient selective nature of HAPs [71][72]. Similar to hypoxic cell radiosensitizers, in order to overcome the barriers associated with HAPs, researchers have begun to explore drug delivery systems that integrate nanotechnology and cellular manipulation [73]. Particularly, researchers have been investigating methods to improve the delivery of HAPs to hypoxic regions of a tumour due to their inefficient selective capability through macrophage-mediated delivery systems [73]. Macrophages serve as an attractive vessel for delivering HAPs to hypoxic tumour microenvironments relative to typical drug administration for three main reasons [74]. One reason includes the ability of macrophages to migrate to hypoxic regions of a tumour through chemoattractant gradients [74][75][76][77]. Additionally, macrophages are able to recognize and clear foreign bodies within the bloodstream, which indicates the ability to uptake nanoparticles; thirdly, macrophages have the ability to target various diseases such as cancer [74][75][76][77]. Furthermore, Yu et al. [74] state that macrophages accumulate within hypoxic regions and are activated by intracellular conditions leading to the release of the contents being withheld, such as HAPs. Thus, the innate ability for macrophages to target hypoxic tumour microenvironments relative to normal tissue, demonstrates a powerful approach to combating tumour hypoxia. Favourable biological structural components in relation to drug delivery are outlined further in this paper.
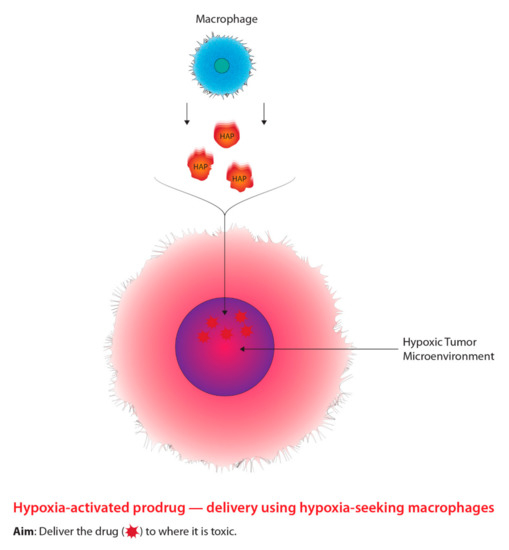
Figure 3. The concept underlying the targeting of hypoxic cells using prodrugs tagged to tumour-seeking macrophages.
3.2. Mechanism of Action
As stated earlier, current research has been focused on advancing drug delivery of HAPs to hypoxic tumour regions mediated by macrophages due to the possibility of HAPs entering normoxic cells and causing detrimental effects on the cell. Macrophages are responsible for the production of inflammatory and antimicrobial cytokines, as well as the removal of pathogens, and are the predominant phagocyte within the immune system [78]. Depending on the environment and host, macrophage regulation can elicit two distinct phenotypes, each with various functionality, M1 macrophages and M2 macrophages, which are further subdivided into M2a, M2b, M2c, and M2d [78]. M1 macrophages are classically activated and generate inflammatory cytokines that inhibit growth, namely, TNF-α and interleukin (IL)-1 [78]. Contrastingly, M2 macrophages are not activated like M1 macrophages and produce anti-inflammatory cytokines and serve as tumour growth promoters [78]. In addition, the four subdivisions of M2 macrophages exhibit various levels of transcriptional changes depending on the stimuli applied [78]. Furthermore, macrophages have the ability to shift between M1 or M2 phenotypes depending on environmental cues [78][79][80]. In the non-targeted field, this was recognized early on [81][82], and it was also found that there was an important correlation between the M2 phenotype, which is correlated with progression to genomic instability and a radioresistant pattern of response, while M1 phenotype is correlated with an apoptotic response to radiation and with a radiosensitive response [83][84][85]. Consideration of NTE in this context might improve treatment outcomes due to a more complete understanding of the mechanisms involved. Interestingly, various diseases, including cancer, demonstrate a disproportionate amount of M1 and M2 macrophage populations [78]. However, within tumours, macrophages are known as tumour-associated macrophages (TAMs), which constitute roughly half of the immune cell populations in tumours and are active during different stages of tumour progression [78][86]. Despite the various phenotype demonstrated by TAMs, overall, these groups of macrophages are classified as M2 macrophages due to the similar responses elicited, such as generating anti-inflammatory cytokines, aids in tumour development, angiogenesis, metastasis, and suppressing the immune system [78][87][88]. Zhang et al. [89] suggest that increased numbers of TAMs, in addition to a high M2:M1 ratio, leads to poor outcomes for various types of cancer. In relation to the poor vasculature system developed by growing tumours, inevitably leading to tumour hypoxia, studies have displayed that TAMs accumulate within hypoxic tumour microenvironments [78]. The migration and infiltration of TAMs are mediated by VEGF and HIF-1. Once entered into the hypoxic region, low intracellular oxygen levels lead to the down-regulation of C-C chemokines, thus rendering TAMs immobile in hypoxic regions [78].
In order to use macrophage-mediated delivery for HAPs, the process requires four distinct phases; “cargo” loading (i.e., whereby cargo refers to the material acquired by macrophages, such as HAPs), maintaining cargo integrity, motility of macrophage in vivo to the target site, and cargo expulsion [73]. In order for macrophages to uptake HAPs, a protective nanoparticle must be encapsulating the drug to prevent the degradation of the drug from intracellular enzymatic conditions induced by macrophages and to protect the macrophage from the drug [78]. For macrophages to uptake a nanoparticle encasing a drug, several characteristics of the nanoparticle surface must be considered for adsorption to macrophage proteins [90]. The three main nanoparticle attributes include curvature, topography, and surface energy; however, other attributes may exist but have yet to be uncovered [90]. Moreover, considerations regarding the ability of macrophage receptors, recognizing these nanoparticles and mechanism of uptake is critical for the drug delivery process [90]. Following uptake of the nanoparticle encasing the drug, it is critical to maintain the integrity of the drug. The drug stability depends on intracellular traffic, particularly the avoidance of lysosomes, due to the potential degradation of the encased drug [91][92]. To prevent cargo degradation, Batrakova et al. [91] suggest that positively charged block-copolymer inhibits lysosome degradation, thus maintaining the stability of the drug. Alternatively, the “backpack” approach is an extreme method for preserving intracellular drug stability, whereby the drug is attached to the surface of a cell carrier [91]. Despite the potential for maintaining drug stability, Batrakova et al. [91] suggest several restraints associated with the “backpack” method, including reduced drug loading, impaired drug release at the target site, and an increase in toxicity and immunogenicity. Consequently, the migration of macrophages is orchestrated through the innate homing properties of macrophages that allow them to travel to hypoxic microenvironments of a tumour [91]. Although the mechanisms related to drug unloading within macrophages continue to be uncovered, several pathways have been hypothesized [91]. One hypothesis suggests that increased concentrations of intracellular calcium is thought to trigger drug release from macrophages [91][93].
Following the release of HAPs from a macrophage carrier, HAPs undergo a series of chemical reactions to activate the drug. Moreover, HAPs display inefficient selective behaviour, which is apparent in one of the most extensively studied HAPs, TPZ [94]. In principle, HAPs are masked or deactivated cytotoxins that are subjected to biotransformations, which are then proceeded by reductive metabolism orchestrated by intracellular oxidoreductases to produce an active compound [95]. Initially, compounds remain inactive due to the positioning of a bioreductive protecting group, which is released following reduction and fragmentation [96][97]. As described by Guise et al. [95], normally, the aforementioned processes are inhibited within normoxic cells due to the levels of intracellular oxygen. However, the significantly lower oxygen concentration is ideal for HAP activation within hypoxic tumour cells [95]. Furthermore, the activation of HAPs can occur in two different manners, through one-electron oxidoreductases to catalyze oxygen-sensitive HAPs or through two-electron oxidoreductases to catalyze oxygen-insensitive HAPs [95]. In regard to oxygen-sensitive HAPs, one-electron oxidoreductases will produce free radicals that can easily be reoxidized into the inactive precursor form, creating a futile metabolic cycle, thus, limiting these HAPs to hypoxic regions [95][98]. Contrastingly, as described by Guise et al. [95], processes using two-electron oxidoreductases are irreversible and are unable to produce oxygen-sensitive radical intermediates; thus, the compound can potentially situate within normoxic and hypoxic tissues, overall, creating HAP activation independent of oxygen concentration. Despite the role of one-electron oxidoreductases and two-electron oxidoreductases in relation to HAP activation, the expression and frequency of these enzymes remain to be elucidated within human tumours [95].
4. Autophagy and Tumour Metabolism
4.1. Background
Another method is the use of drugs that target tumour metabolism. Tumours demonstrate a growth advantage through a shift in metabolism, specifically, from oxidative phosphorylation to glycolytic metabolism, which is driven by HIF-1–pyruvate dehydrogenase kinase 1 (PDK1) [99]. Consequently, the shift in metabolism causes tumours to conserve oxygen supplies and induces a compensatory response by increasing glycolysis through the reduction of mitochondrial processes [99]. Moreover, the inhibition of PDK leads to an increase in tumour hypoxia [99]. Researchers have demonstrated that dichloroacetate (DCA) acts as a PDK inhibitor, increasing mitochondrial functioning within tumours, thus, resuming oxidative metabolism similar to normal tissues [100]. Aside from targeting the transition from oxidative phosphorylation to glycolytic metabolism, other cancer therapeutics targeting autophagy, another molecular process aiding in tumour metabolism, has been garnering recent attention. The overall concept for this approach is outlined in Figure 4. The process of autophagy in relation to cancer involves the degradation of damaged cellular components that are recycled to meet the metabolic demands of cancer cells [101]. Mizushima and Komatsu [102] suggest that low baseline levels of autophagy are essential for preventing toxicity in tissues by preventing the build-up of damaged proteins and organelles. Autophagy is characterized as a “double-edged sword”, primarily due to its dual role in tumourigenesis serving as a tumour suppressor and tumour promotor, depending on the type of tissue and stage of the tumour [101]. Several studies on human prostate, breast, and ovarian cancers, displayed partial monoallelic loss in one essential autophagy gene, ATG6/Beclin-1 [101][103][104][105]. Furthermore, the impairment of proper autophagy functioning in tumours serves as a signal for identifying cancer [106]. Research has also suggested that autophagy acts as a tumour promoter due to growth enhancement and survival capabilities [107]. As previously mentioned, hypoxic tumour environments are severely lacking in metabolic requirements due to primitive vasculature systems; however, autophagy serves to meet metabolic demands through the recycling of intracellular components [107][108][109]. Normally, when damaged or old cells are removed through autophagy, there is a release of structural biological components, such as amino acids, nucleotides, and fatty acids [110]. Furthermore, these intracellular components can be recycled and used for tumour metabolic demands; however, suppressing autophagy through the partial deletion of the Beclin-1 gene leads to increased cell death [107]. Current preclinical studies have determined that inhibiting autophagy has improved cancer patient outcomes [111]. At present, the only autophagy inhibiting drugs viable for clinical studies are chloroquine (CQ) and hydroxychloroquine (HCQ), a derivative of CQ.
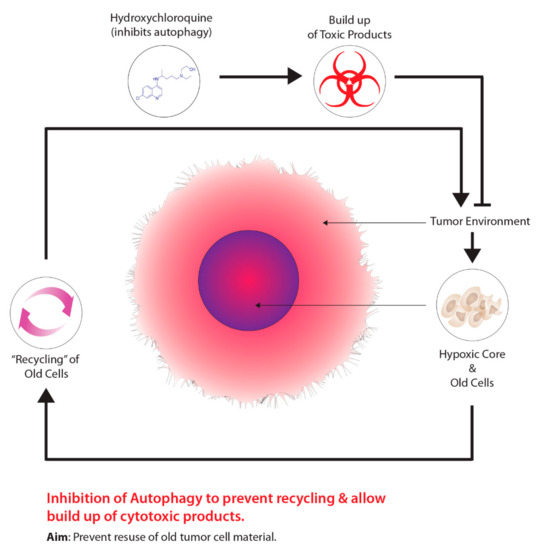
Figure 4. Harnessing autophagy to destroy hypoxic cells.
4.2. Mechanisms of Action
As previously mentioned, recent studies on tumour metabolism have evolved from analyzing the shift in oxidative to glycolytic processes within tumours to the role of autophagy in tumour progression and the subsequent development of autophagy inhibitors. In particular, the most notable autophagy inhibitors are CQ and HCQ, which are classified as 4-aminoquinoline agents and were first intended as anti-malarial drugs [110]. HCQ is a derivative of CQ and is distinguished by the addition of a hydroxyl group on the beta carbon of the tertiary amino ethyl situated on the terminus side of the quinolone base [112]. Furthermore, the addition of hydroxyl group restricts the movement of HCQ across blood-retinal barriers, overall resulting in lower toxicity relative to CQ [110][112][113]. As stated earlier, autophagy has two distinct roles in relation to tumourigenesis, namely as a tumour suppressor or tumour promoter depending on the stage of tumour development and the tissue type [110]. During the early stage of tumour development, autophagy acts as a tumour suppressor due to its ability to clear defective cells, thus, maintaining cell homeostasis [110][114][115]. Moreover, various proteins associated with autophagy that directly suppress tumour development include Beclin-1, UVRAG, and Bif-1, as well as components that destroy proteins associated with tumour growth such as p62/SQSTM1 [110][116]. Contrastingly, during the later stages of tumourigenesis, levels of autophagy increase and act as a tumour promoter in response to harsh intracellular environments such as starvation, hypoxia, and organelle damage [110]. Furthermore, one major characteristic of autophagy is the ability to recycle nutrients, which can be used to sustain tumour development [110]. Moreover, increased autophagy activity is associated with the destruction of cell growth regulators, as well as suppression of DNA damage mechanisms [110][114][115][116]. In order to combat autophagy, CQ and HCQ act as inhibitors during the late stages of autophagy, particularly when lysosomes and autophagosomes fuse together [110]. Townsend et al. [112] state that when CQ or HCQ enter the lysosome, this causes the protonation of these compounds, ultimately trapping these compounds within the acidic lysosome environment, causing the inhibition of lysosome degradation enzymes. Lysosomes are integral during autophagy, as these cells are responsible for the degradation of macromolecules that can be reused within cells [117]. Thus, cells treated with CQ or HCQ cannot undergo lysosomal digestion [112][118]. Moreover, preventing the proper lysosome functioning, in turn, prevents the supply of macromolecules required for tumour growth, thus, serving as an attractive method for targeting tumour metabolism relative to normal tissue [110].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22168651
References
- Ogawa, K.; Kohshi, K.; Ishiuchi, S.; Matsushita, M.; Yoshimi, N.; Murayama, S. Old but New Methods in Radiation Oncology: Hyperbaric Oxygen Therapy. Int. J. Clin. Oncol. 2013, 18, 364–370.
- Jain, K.K. Textbook of Hyperbaric Medicine; Springer International Publishing: Cham, Switzerland, 1953; ISBN 978-3-319-47138-9.
- Hartmann, K.A.; van der Kleij, A.J.; Carl, U.M.; Hulshof, M.C.; Willers, R.; Sminia, P. Effects of Hyperbaric Oxygen and Normobaric Carbogen on the Radiation Response of the Rat Rhabdomyosarcoma R1H. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 1037–1044.
- Hall, E.J.; Giaccia, A.J. Radiobiology for the Radiologist, 7th ed.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012; ISBN 978-1-60831-193-4.
- Churchill-Davidson, I. The Oxygen Effect in Radiotherapy—Historical Review. Hyperb. Oxyg. Radiat. Ther. Cancer 1968, 1, 1–15.
- Bennett, M.H.; Feldmeier, J.; Smee, R.; Milross, C. Hyperbaric Oxygenation for Tumour Sensitisation to Radiotherapy. Cochrane Database Syst. Rev. 2018, 2018, CD005007.
- Kohshi, K.; Kinoshita, Y.; Imada, H.; Kunugita, N.; Abe, H.; Terashima, H.; Tokui, N.; Uemura, S. Effects of Radiotherapy after Hyperbaric Oxygenation on Malignant Gliomas. Br. J. Cancer 1999, 80, 236–241.
- Moen, I.; Stuhr, L.E.B. Hyperbaric Oxygen Therapy and Cancer—A Review. Target. Oncol. 2012, 7, 233–242.
- Kjellen, E.; Joiner, M.C.; Collier, J.M.; Johns, H.; Rojas, A. A Therapeutic Benefit from Combining Normobaric Carbogen or Oxygen with Nicotinamide in Fractionated X-Ray Treatments. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 1991, 22, 81–91.
- Tran, L.-B.-A.; Bol, A.; Labar, D.; Karroum, O.; Bol, V.; Jordan, B.; Grégoire, V.; Gallez, B. Potential Role of Hypoxia Imaging Using 18F-FAZA PET to Guide Hypoxia-Driven Interventions (Carbogen Breathing or Dose Escalation) in Radiation Therapy. Radiother. Oncol. 2014, 113, 204–209.
- Harrison, L.B.; Chadha, M.; Hill, R.J.; Hu, K.; Shasha, D. Impact of Tumour Hypoxia and Anemia on Radiation Therapy Outcomes. Oncologist 2002, 7, 492–508.
- Fleming, I.N.; Manavaki, R.; Blower, P.J.; West, C.; Williams, K.J.; Harris, A.L.; Domarkas, J.; Lord, S.; Baldry, C.; Gilbert, F.J. Imaging Tumour Hypoxia with Positron Emission Tomography. Br. J. Cancer 2015, 112, 238–250.
- Unterrainer, M.; Eze, C.; Ilhan, H.; Marschner, S.; Roengvoraphoj, O.; Schmidt-Hegemann, N.S.; Walter, F.; Kunz, W.G.; af Rosenschöld, P.M.; Jeraj, R.; et al. Recent Advances of PET Imaging in Clinical Radiation Oncology. Radiat. Oncol. 2020, 15, 88.
- Chapman, J.D. Hypoxic Sensitizers—Implications for Radiation Therapy. N. Engl. J. Med. 1979, 301, 1429–1432.
- Nunn, A.; Linder, K.; Strauss, H.W. Nitroimidazoles and Imaging Hypoxia. Eur. J. Nucl. Med. 1995, 22, 265–280.
- Edwards, D.I. Nitroimidazole Drugs--Action and Resistance Mechanisms. I. Mechanisms of Action. J. Antimicrob. Chemother. 1993, 31, 9–20.
- Lopci, E.; Grassi, I.; Chiti, A.; Nanni, C.; Cicoria, G.; Toschi, L.; Fonti, C.; Lodi, F.; Mattioli, S.; Fanti, S. PET Radiopharmaceuticals for Imaging of Tumour Hypoxia: A Review of the Evidence. Am. J. Nucl. Med. Mol. Imaging 2014, 4, 365–384.
- Bourgeois, M.; Rajerison, H.; Guerard, F.; Mougin-Degraef, M.; Barbet, J.; Michel, N.; Cherel, M.; Faivre-Chauvet, A. Contribution of [64Cu]-ATSM PET in Molecular Imaging of Tumour Hypoxia Compared to Classical [18F]-MISO—A Selected Review. Nucl. Med. Rev. Cent. East. Eur. 2011, 14, 90–95.
- Hoigebazar, L.; Jeong, J.M. Hypoxia Imaging Agents Labeled with Positron Emitters. Recent Results Cancer Res. Fortschr. Krebsforsch. Prog. Dans Rech. Sur Cancer 2013, 194, 285–299.
- Takasawa, M.; Moustafa, R.R.; Baron, J.-C. Applications of Nitroimidazole in Vivo Hypoxia Imaging in Ischemic Stroke. Stroke 2008, 39, 1629–1637.
- Kumar, P.; Stypinski, D.; Xia, H.; McEwan, A.J.B.; Machulla, H.-J.; Wiebe, L.I. Fluoroazomycin Arabinoside (FAZA): Synthesis, 2H and 3H-Labelling and Preliminary Biological Evaluation of a Novel 2-Nitroimidazole Marker of Tissue Hypoxia. J. Label. Compd. Radiopharm. 1999, 42, 3–16.
- Busk, M.; Horsman, M.R.; Jakobsen, S.; Bussink, J.; van der Kogel, A.; Overgaard, J. Cellular Uptake of PET Tracers of Glucose Metabolism and Hypoxia and Their Linkage. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 2294–2303.
- Quartuccio, N.; Laudicella, R.; Mapelli, P.; Guglielmo, P.; Pizzuto, D.A.; Boero, M.; Arnone, G.; Picchio, M. Young AIMN Working Group. Hypoxia PET Imaging beyond 18F-FMISO in Patients with High-Grade Glioma: 18F-FAZA and Other Hypoxia Radiotracers. Clin. Transl. Imaging 2020, 8, 11–20.
- Busk, M.; Mortensen, L.S.; Nordsmark, M.; Overgaard, J.; Jakobsen, S.; Hansen, K.V.; Theil, J.; Kallehauge, J.F.; D’Andrea, F.P.; Steiniche, T.; et al. PET Hypoxia Imaging with FAZA: Reproducibility at Baseline and during Fractionated Radiotherapy in Tumour-Bearing Mice. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 186–197.
- Adams, G.E.; Flockhart, I.R.; Smithen, C.E.; Stratford, I.J.; Wardman, P.; Watts, M.E. Electron-Affinic Sensitization. VII. A Correlation between Structures, One-Electron Reduction Potentials, and Efficiencies of Nitroimidazoles as Hypoxic Cell Radiosensitizers. Radiat. Res. 1976, 67, 9–20.
- Wardman, P. Chemical Radiosensitizers for Use in Radiotherapy. Clin. Oncol. 2007, 19, 397–417.
- Hall, E.J. Radiobiology for the Radiologist, 5th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2000; ISBN 978-0-7817-2649-8.
- Overgaard, J.; Sand Hansen, H.; Lindeløv, B.; Overgaard, M.; Jørgensen, K.; Rasmusson, B.; Berthelsen, A. Nimorazole as a Hypoxic Radiosensitizer in the Treatment of Supraglottic Larynx and Pharynx Carcinoma. First Report from the Danish Head and Neck Cancer Study (DAHANCA) Protocol 5-85. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 1991, 20 (Suppl. S1), 143–149.
- Overgaard, J.; Hansen, H.S.; Overgaard, M.; Bastholt, L.; Berthelsen, A.; Specht, L.; Lindeløv, B.; Jørgensen, K. A Randomized Double-Blind Phase III Study of Nimorazole as a Hypoxic Radiosensitizer of Primary Radiotherapy in Supraglottic Larynx and Pharynx Carcinoma. Results of the Danish Head and Neck Cancer Study (DAHANCA) Protocol 5-85. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 1998, 46, 135–146.
- Graham, K.; Unger, E. Overcoming Tumour Hypoxia as a Barrier to Radiotherapy, Chemotherapy and Immunotherapy in Cancer Treatment. Int. J. Nanomed. 2018, 13, 6049–6058.
- Teicher, B.A.; Lazo, J.S.; Sartorelli, A.C. Classification of Antineoplastic Agents by Their Selective Toxicities toward Oxygenated and Hypoxic Tumour Cells. Cancer Res. 1981, 41, 73–81.
- Wardman, P. Nitroimidazoles as Hypoxic Cell Radiosensitizers and Hypoxia Probes: Misonidazole, Myths and Mistakes. Br. J. Radiol. 2019, 92, 20170915.
- Wang, H.; Mu, X.; He, H.; Zhang, X.-D. Cancer Radiosensitizers. Trends Pharmacol. Sci. 2018, 39, 24–48.
- Matsumura, Y.; Maeda, H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumouritropic Accumulation of Proteins and the Antitumour Agent Smancs. Cancer Res. 1986, 46, 6387–6392.
- Wong, A.D.; Ye, M.; Ulmschneider, M.B.; Searson, P.C. Quantitative Analysis of the Enhanced Permeation and Retention (EPR) Effect. PLoS ONE 2015, 10, e0123461.
- Minchinton, A.I.; Tannock, I.F. Drug Penetration in Solid Tumours. Nat. Rev. Cancer 2006, 6, 583–592.
- Golombek, S.K.; May, J.-N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumour Targeting via EPR: Strategies to Enhance Patient Responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38.
- Torchilin, V. Tumour Delivery of Macromolecular Drugs Based on the EPR Effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135.
- Prabhakar, U.; Maeda, H.; Jain, R.K.; Sevick-Muraca, E.M.; Zamboni, W.; Farokhzad, O.C.; Barry, S.T.; Gabizon, A.; Grodzinski, P.; Blakey, D.C. Challenges and Key Considerations of the Enhanced Permeability and Retention (EPR) Effect for Nanomedicine Drug Delivery in Oncology. Cancer Res. 2013, 73, 2412–2417.
- Maeda, H.; Nakamura, H.; Fang, J. The EPR Effect for Macromolecular Drug Delivery to Solid Tumours: Improvement of Tumour Uptake, Lowering of Systemic Toxicity, and Distinct Tumour Imaging in Vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79.
- Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J.W.; Thurston, G.; Roberge, S.; Jain, R.K.; McDonald, D.M. Openings between Defective Endothelial Cells Explain Tumour Vessel Leakiness. Am. J. Pathol. 2000, 156, 1363–1380.
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an Emerging Platform for Cancer Therapy. Nat. Nanotechnol. 2007, 2, 751–760.
- Cheng, Z.; Al Zaki, A.; Hui, J.Z.; Muzykantov, V.R.; Tsourkas, A. Multifunctional Nanoparticles: Cost versus Benefit of Adding Targeting and Imaging Capabilities. Science 2012, 338, 903–910.
- Her, S.; Jaffray, D.A.; Allen, C. Gold Nanoparticles for Applications in Cancer Radiotherapy: Mechanisms and Recent Advancements. Adv. Drug Deliv. Rev. 2017, 109, 84–101.
- Mi, Y.; Shao, Z.; Vang, J.; Kaidar-Person, O.; Wang, A.Z. Application of Nanotechnology to Cancer Radiotherapy. Cancer Nanotechnol. 2016, 7, 11.
- Babaei, M.; Ganjalikhani, M. The Potential Effectiveness of Nanoparticles as Radio Sensitizers for Radiotherapy. BioImpacts BI 2014, 4, 15–20.
- Chen, Y.; Yang, J.; Fu, S.; Wu, J. Gold Nanoparticles as Radiosensitizers in Cancer Radiotherapy. Int. J. Nanomed. 2020, 15, 9407–9430.
- Dimitriou, N.M.; Tsekenis, G.; Balanikas, E.C.; Pavlopoulou, A.; Mitsiogianni, M.; Mantso, T.; Pashos, G.; Boudouvis, A.G.; Lykakis, I.N.; Tsigaridas, G.; et al. Gold Nanoparticles, Radiations and the Immune System: Current Insights into the Physical Mechanisms and the Biological Interactions of This New Alliance towards Cancer Therapy. Pharmacol. Ther. 2017, 178, 1–17.
- Retif, P.; Pinel, S.; Toussaint, M.; Frochot, C.; Chouikrat, R.; Bastogne, T.; Barberi-Heyob, M. Nanoparticles for Radiation Therapy Enhancement: The Key Parameters. Theranostics 2015, 5, 1030–1044.
- Yao, X.; Huang, C.; Chen, X.; Yi, Z.; Sanche, L. Chemical Radiosensitivity of DNA Induced by Gold Nanoparticles. J. Biomed. Nanotechnol. 2015, 11, 478–485.
- Liu, K.; Han, L.; Zhuang, J.; Yang, D.-P. Protein-Directed Gold Nanoparticles with Excellent Catalytic Activity for 4-Nitrophenol Reduction. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 78, 429–434.
- Mateo, D.; Morales, P.; Ávalos, A.; Haza, A.I. Oxidative Stress Contributes to Gold Nanoparticle-Induced Cytotoxicity in Human Tumour Cells. Toxicol. Mech. Methods 2014, 24, 161–172.
- Decrock, E.; Hoorelbeke, D.; Ramadan, R.; Delvaeye, T.; De Bock, M.; Wang, N.; Krysko, D.V.; Baatout, S.; Bultynck, G.; Aerts, A.; et al. Calcium, Oxidative Stress and Connexin Channels, a Harmonious Orchestra Directing the Response to Radiotherapy Treatment? Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1099–1120.
- Sinclair, W.K.; Morton, R.A. X-Ray Sensitivity during the Cell Generation Cycle of Cultured Chinese Hamster Cells. Radiat. Res. 1966, 29, 450–474.
- Mackey, M.A.; Saira, F.; Mahmoud, M.A.; El-Sayed, M.A. Inducing Cancer Cell Death by Targeting Its Nucleus: Solid Gold Nanospheres versus Hollow Gold Nanocages. Bioconjug. Chem. 2013, 24, 897–906.
- Cui, L.; Tse, K.; Zahedi, P.; Harding, S.M.; Zafarana, G.; Jaffray, D.A.; Bristow, R.G.; Allen, C. Hypoxia and Cellular Localization Influence the Radiosensitizing Effect of Gold Nanoparticles (AuNPs) in Breast Cancer Cells. Radiat. Res. 2014, 182, 475–488.
- Pan, Y.; Leifert, A.; Ruau, D.; Neuss, S.; Bornemann, J.; Schmid, G.; Brandau, W.; Simon, U.; Jahnen-Dechent, W. Gold Nanoparticles of Diameter 1.4 Nm Trigger Necrosis by Oxidative Stress and Mitochondrial Damage. Small 2009, 5, 2067–2076.
- Butterworth, K.T.; Coulter, J.A.; Jain, S.; Forker, J.; McMahon, S.J.; Schettino, G.; Prise, K.M.; Currell, F.J.; Hirst, D.G. Evaluation of Cytotoxicity and Radiation Enhancement Using 1.9 Nm Gold Particles: Potential Application for Cancer Therapy. Nanotechnology 2010, 21, 295101.
- Liu, Y.; Zhang, P.; Li, F.; Jin, X.; Li, J.; Chen, W.; Li, Q. Metal-Based NanoEnhancers for Future Radiotherapy: Radiosensitizing and Synergistic Effects on Tumour Cells. Theranostics 2018, 8, 1824–1849.
- Djuzenova, C.S.; Elsner, I.; Katzer, A.; Worschech, E.; Distel, L.V.; Flentje, M.; Polat, B. Radiosensitivity in Breast Cancer Assessed by the Histone γ-H2AX and 53BP1 Foci. Radiat. Oncol. 2013, 8, 98.
- Vignard, J.; Mirey, G.; Salles, B. Ionizing-Radiation Induced DNA Double-Strand Breaks: A Direct and Indirect Lighting Up. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2013, 108, 362–369.
- Mothersill, C.; Crean, M.; Lyons, M.; McSweeney, J.; Mooney, R.; O’Reilly, J.; Seymour, C.B. Expression of Delayed Toxicity and Lethal Mutations in the Progeny of Human Cells Surviving Exposure to Radiation and other Environmental Mutagens. Int. J. Radiat. Biol. 1998, 74, 673–680.
- Glaviano, A.; Nayak, V.; Cabuy, E.; Baird, D.M.; Yin, Z.; Newson, R.; Ladon, D.; Rubio, M.A.; Slijepcevic, P.; Lyng, F.; et al. Effects of HTERT on Metal Ion-Induced Genomic Instability. Oncogene 2006, 25, 3424–3435.
- Glaviano, A.; Mothersill, C.; Case, C.P.; Rubio, M.A.; Newson, R.; Lyng, F. Effects of HTERT on Genomic Instability Caused by Either Metal or Radiation or Combined Exposure. Mutagenesis 2009, 24, 25–33.
- Coen, N.; Kadhim, M.A.; Wright, E.G.; Case, C.P.; Mothersill, C.E. Particulate Debris from a Titanium Metal Prosthesis Induces Genomic Instability in Primary Human Fibroblast Cells. Br. J. Cancer 2003, 88, 548–552.
- Rosa, S.; Connolly, C.; Schettino, G.; Butterworth, K.T.; Prise, K.M. Biological Mechanisms of Gold Nanoparticle Radiosensitization. Cancer Nanotechnol. 2017, 8, 2.
- Ghita, M.; McMahon, S.J.; Taggart, L.E.; Butterworth, K.T.; Schettino, G.; Prise, K.M. A Mechanistic Study of Gold Nanoparticle Radiosensitisation Using Targeted Microbeam Irradiation. Sci. Rep. 2017, 7, 44752.
- Brown, J.M.; Siim, B.G. Hypoxia-Specific Cytotoxins in Cancer Therapy. Semin. Radiat. Oncol. 1996, 6, 22–36.
- Zeng, Y.; Ma, J.; Zhan, Y.; Xu, X.; Zeng, Q.; Liang, J.; Chen, X. Hypoxia-Activated Prodrugs and Redox-Responsive Nanocarriers. Int. J. Nanomed. 2018, 13, 6551–6574.
- Rischin, D.; Peters, L.; Fisher, R.; Macann, A.; Denham, J.; Poulsen, M.; Jackson, M.; Kenny, L.; Penniment, M.; Corry, J.; et al. Tirapazamine, Cisplatin, and Radiation versus Fluorouracil, Cisplatin, and Radiation in Patients with Locally Advanced Head and Neck Cancer: A Randomized Phase II Trial of the Trans-Tasman Radiation Oncology Group (TROG 98.02). J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 79–87.
- Thambi, T.; Park, J.H.; Lee, D.S. Hypoxia-Responsive Nanocarriers for Cancer Imaging and Therapy: Recent Approaches and Future Perspectives. Chem. Commun. 2016, 52, 8492–8500.
- Gao, G.H.; Li, Y.; Lee, D.S. Environmental PH-Sensitive Polymeric Micelles for Cancer Diagnosis and Targeted Therapy. J. Control. Release 2013, 169, 180–184.
- Visser, J.G.; Van Staden, A.D.P.; Smith, C. Harnessing Macrophages for Controlled-Release Drug Delivery: Lessons From Microbes. Front. Pharmacol. 2019, 10, 22.
- Yu, H.; Yang, Z.; Li, F.; Xu, L.; Sun, Y. Cell-Mediated Targeting Drugs Delivery Systems. Drug Deliv. 2020, 27, 1425–1437.
- Dong, X.; Chu, D.; Wang, Z. Leukocyte-Mediated Delivery of Nanotherapeutics in Inflammatory and Tumour Sites. Theranostics 2017, 7, 751–763.
- Pierigè, F.; Serafini, S.; Rossi, L.; Magnani, M. Cell-Based Drug Delivery. Adv. Drug Deliv. Rev. 2008, 60, 286–295.
- Yousefpour, P.; Chilkoti, A. Co-Opting Biology to Deliver Drugs. Biotechnol. Bioeng. 2014, 111, 1699–1716.
- Wang, H.-F.; Liu, Y.; Yang, G.; Zhao, C.-X. Macrophage-Mediated Cancer Drug Delivery. Mater. Today Sustain. 2021, 11–12, 100055.
- Liu, X.; Li, J.; Peng, X.; Lv, B.; Wang, P.; Zhao, X.; Yu, B. Geraniin Inhibits LPS-Induced THP-1 Macrophages Switching to M1 Phenotype via SOCS1/NF-ΚB Pathway. Inflammation 2016, 39, 1421–1433.
- Zhu, Y.; Li, X.; Chen, J.; Chen, T.; Shi, Z.; Lei, M.; Zhang, Y.; Bai, P.; Li, Y.; Fei, X. The Pentacyclic Triterpene Lupeol Switches M1 Macrophages to M2 and Ameliorates Experimental Inflammatory Bowel Disease. Int. Immunopharmacol. 2016, 30, 74–84.
- Ponnaiya, B.; Cornforth, M.N.; Ullrich, R.L. Radiation-Induced Chromosomal Instability in BALB/c and C57BL/6 Mice: The Difference Is as Clear as Black and White. Radiat. Res. 1997, 147, 121–125.
- Mothersill, C.; Rea, D.; Wright, E.G.; Lorimore, S.A.; Murphy, D.; Seymour, C.B.; O’Malley, K. Individual Variation in the Production of a “bystander Signal” Following Irradiation of Primary Cultures of Normal Human Urothelium. Carcinogenesis 2001, 22, 1465–1471.
- Lindsay, K.J.; Coates, P.J.; Lorimore, S.A.; Wright, E.G. The Genetic Basis of Tissue Responses to Ionizing Radiation. Br. J. Radiol. 2007, 80, S2–S6.
- Lorimore, S.A.; Mukherjee, D.; Robinson, J.I.; Chrystal, J.A.; Wright, E.G. Long-Lived Inflammatory Signaling in Irradiated Bone Marrow Is Genome Dependent. Cancer Res. 2011, 71, 6485–6491.
- Mukherjee, D.; Coates, P.J.; Lorimore, S.A.; Wright, E.G. Responses to Ionizing Radiation Mediated by Inflammatory Mechanisms. J. Pathol. 2014, 232, 289–299.
- Poh, A.R.; Ernst, M. Targeting Macrophages in Cancer: From Bench to Bedside. Front. Oncol. 2018, 8, 49.
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumour-Associated Macrophages as Major Players in the Tumour Microenvironment. Cancers 2014, 6, 1670–1690.
- Aras, S.; Zaidi, M.R. TAMeless Traitors: Macrophages in Cancer Progression and Metastasis. Br. J. Cancer 2017, 117, 1583–1591.
- Zhang, M.; He, Y.; Sun, X.; Li, Q.; Wang, W.; Zhao, A.; Di, W. A High M1/M2 Ratio of Tumour-Associated Macrophages Is Associated with Extended Survival in Ovarian Cancer Patients. J. Ovarian Res. 2014, 7, 19.
- Gustafson, H.H.; Holt-Casper, D.; Grainger, D.W.; Ghandehari, H. Nanoparticle Uptake: The Phagocyte Problem. Nano Today 2015, 10, 487–510.
- Batrakova, E.V.; Gendelman, H.E.; Kabanov, A.V. Cell-Mediated Drugs Delivery. Expert Opin. Drug Deliv. 2011, 8, 415–433.
- Nowacek, A.S.; Miller, R.L.; McMillan, J.; Kanmogne, G.; Kanmogne, M.; Mosley, R.L.; Ma, Z.; Graham, S.; Chaubal, M.; Werling, J.; et al. NanoART Synthesis, Characterization, Uptake, Release and Toxicology for Human Monocyte-Macrophage Drug Delivery. Nanomedicine 2009, 4, 903–917.
- Söllner, T.; Bennett, M.K.; Whiteheart, S.W.; Scheller, R.H.; Rothman, J.E. A Protein Assembly-Disassembly Pathway in Vitro That May Correspond to Sequential Steps of Synaptic Vesicle Docking, Activation, and Fusion. Cell 1993, 75, 409–418.
- Moriwaki, T.; Okamoto, S.; Sasanuma, H.; Nagasawa, H.; Takeda, S.; Masunaga, S.-I.; Tano, K. Cytotoxicity of Tirapazamine (3-Amino-1,2,4-Benzotriazine-1,4-Dioxide)-Induced DNA Damage in Chicken DT40 Cells. Chem. Res. Toxicol. 2017, 30, 699–704.
- Guise, C.P.; Mowday, A.M.; Ashoorzadeh, A.; Yuan, R.; Lin, W.-H.; Wu, D.-H.; Smaill, J.B.; Patterson, A.V.; Ding, K. Bioreductive Prodrugs as Cancer Therapeutics: Targeting Tumour Hypoxia. Chin. J. Cancer 2014, 33, 80–86.
- Mistry, I.N.; Thomas, M.; Calder, E.D.D.; Conway, S.J.; Hammond, E.M. Clinical Advances of Hypoxia-Activated Prodrugs in Combination With Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2017, 98, 1183–1196.
- O’Connor, L.J.; Cazares-Körner, C.; Saha, J.; Evans, C.N.G.; Stratford, M.R.L.; Hammond, E.M.; Conway, S.J. Design, Synthesis and Evaluation of Molecularly Targeted Hypoxia-Activated Prodrugs. Nat. Protoc. 2016, 11, 781–794.
- Stratford, I.J.; Workman, P. Bioreductive Drugs into the next Millennium. Anticancer. Drug Des. 1998, 13, 519–528.
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 Mediates Adaptation to Hypoxia by Actively Downregulating Mitochondrial Oxygen Consumption. Cell Metab. 2006, 3, 187–197.
- Cairns, R.A.; Papandreou, I.; Sutphin, P.D.; Denko, N.C. Metabolic Targeting of Hypoxia and HIF1 in Solid Tumours Can Enhance Cytotoxic Chemotherapy. Proc. Natl. Acad. Sci. USA 2007, 104, 9445–9450.
- White, E. The Role for Autophagy in Cancer. J. Clin. Investig. 2015, 125, 42–46.
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741.
- Aita, V.M.; Liang, X.H.; Murty, V.V.; Pincus, D.L.; Yu, W.; Cayanis, E.; Kalachikov, S.; Gilliam, T.C.; Levine, B. Cloning and Genomic Organization of Beclin 1, a Candidate Tumour Suppressor Gene on Chromosome 17q21. Genomics 1999, 59, 59–65.
- Choi, A.M.K.; Ryter, S.W.; Levine, B. Autophagy in Human Health and Disease. N. Engl. J. Med. 2013, 368, 651–662.
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of Autophagy and Inhibition of Tumourigenesis by Beclin 1. Nature 1999, 402, 672–676.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674.
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466.
- Rabinowitz, J.D.; White, E. Autophagy and Metabolism. Science 2010, 330, 1344–1348.
- Liu, E.Y.; Ryan, K.M. Autophagy and Cancer--Issues We Need to Digest. J. Cell Sci. 2012, 125, 2349–2358.
- Verbaanderd, C.; Maes, H.; Schaaf, M.B.; Sukhatme, V.P.; Pantziarka, P.; Sukhatme, V.; Agostinis, P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)—Chloroquine and Hydroxychloroquine as Anti-Cancer Agents. Ecancermedicalscience 2017, 11, 781.
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting Autophagy in Cancer. Nat. Rev. Cancer 2017, 17, 528–542.
- Townsend, K.N.; Hughson, L.R.K.; Schlie, K.; Poon, V.I.; Westerback, A.; Lum, J.J. Autophagy Inhibition in Cancer Therapy: Metabolic Considerations for Antitumour Immunity. Immunol. Rev. 2012, 249, 176–194.
- Ben-Zvi, I.; Kivity, S.; Langevitz, P.; Shoenfeld, Y. Hydroxychloroquine: From Malaria to Autoimmunity. Clin. Rev. Allergy Immunol. 2012, 42, 145–153.
- Viry, E.; Paggetti, J.; Baginska, J.; Mgrditchian, T.; Berchem, G.; Moussay, E.; Janji, B. Autophagy: An Adaptive Metabolic Response to Stress Shaping the Antitumour Immunity. Biochem. Pharmacol. 2014, 92, 31–42.
- Cicchini, M.; Karantza, V.; Xia, B. Molecular Pathways: Autophagy in Cancer—A Matter of Timing and Context. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 498–504.
- Cheong, H. Integrating Autophagy and Metabolism in Cancer. Arch. Pharm. Res. 2015, 38, 358–371.
- Yim, W.W.-Y.; Mizushima, N. Lysosome Biology in Autophagy. Cell Discov. 2020, 6, 1–12.
- Tasdemir, E.; Galluzzi, L.; Maiuri, M.C.; Criollo, A.; Vitale, I.; Hangen, E.; Modjtahedi, N.; Kroemer, G. Methods for Assessing Autophagy and Autophagic Cell Death. Methods Mol. Biol. 2008, 445, 29–76.