Gluten-related disorders (GRDs) are a group of diseases that involve the activation of the immune system triggered by the ingestion of gluten, with a worldwide prevalence of 5%. Among them, Celiac disease (CeD) is a T-cell-mediated autoimmune disease causing a plethora of symptoms from diarrhea and malabsorption to lymphoma. Even though GRDs have been intensively studied, the environmental triggers promoting the diverse reactions to gluten proteins in susceptible individuals remain elusive. It has been proposed that pathogens could act as disease-causing environmental triggers of CeD by molecular mimicry mechanisms. Additionally, it could also be possible that unrecognized molecular, structural, and physical parallels between gluten and bacteria have a relevant role.
1. Introduction
Celiac disease (CeD) is a chronic, small-intestinal T-cell-mediated autoimmune disease triggered by the ingestion of dietary gluten from common food grains such as wheat, rye, and barley, in genetically predisposed individuals with a prevalence of about 1% in the general population with regional differences [
1]. Other gluten-related disorders (GRDs) such as non-celiac gluten sensitivity (NCGS) are less understood and have a prevalence rate between 0.5% and 5% [
2]. Nowadays, the only proven treatment for GRDs is strict and life-long adherence to a gluten-free diet [
3].
Viral and bacterial pathogens have long been suspected of triggering immune responses that are directed toward autoimmunity in CeD. In 2002, the group of Khosla showed that the immunodominant gluten fragment has sequence similarity with pertactin, a highly immunogenic protein from the bacterium
Bordetella pertussis; however, this result was not further investigated [
4]. It recently identified and characterized a number of mimics of HLA-DQ2.5-restricted gliadin determinants derived from the commensal bacterium
Pseudomonas fluorescens, activating disease-relevant gliadin reactive T cells isolated from CeD patients [
5]. This report was a major proof of concept that a molecular mimicry mechanism may trigger CeD.
Beyond T-cell activation in CeD, it was also proposed that gluten proteins have functional similarities with non-replicative pathogens such as prions [
6,
7]. The main issue with gluten proteins is that the human digestive proteases can only partially degrade them leading to a mixture of peptides that elicit immune and toxic effects in predisposed individuals and cell lines [
8]. The mononuclear phagocytic system (MPS), composed mainly of macrophages, neutrophils, and dendritic cells, is part of the first line of defense against pathogens. Pathogens are recognized by various immune cells, such as macrophages and dendritic cells, via pathogen-associated molecular patterns (PAMPs) on the pathogen surface, which interact with complementary pattern-recognition receptors (PRRs) on the pathogen surface the immune cell surfaces. While PRR activation is a central component to the resulting immune response, innate cells also respond to the size and shape of the pathogens and the spatial organization of PAMPs on the bacterial surface. In recent years, it has become evident that the MPS can engulf other non-self-systems such as synthetic nanoparticles generating in many cases an immune response against the foreign nanosystem [
9,
10]. From a pathological standpoint, there are some concerns in the possibility that the presence of nanostructures could exacerbate or prolong PRR-driven inflammatory reactions, leading to uncontrolled tissue-damaging inflammation. In this context, it was demonstrated that pepsin digests of gliadin form spontaneously amyloid-like structures that trigger genes in the gut epithelial cell model Caco-2 involved in recruiting specialized immune cells [
11]. Furthermore, the most studied gluten peptides are 33-mer peptides and the p31-43 fragment that form peptide nanostructures, too. Both peptides trigger the adaptive or an innate immune response, respectively, in CeD patients, animal models, and cell lines culture [
12,
13,
14,
15,
16].
The immunodominant 33-mer peptide comprises residues 57 to 89 of α-2-gliadin (LQLQPFPQPQLPYPQPQLPYPQPQLPYPQPQPF). In total, 39% of the 33-mer peptide residues are prolines leading to a type-II polyproline (PPII) conformation in solution, which is known to be bound to MHC class-II molecules [
14]. When the 33-mer accumulates, it forms superstructures ranging from dimers to nano- and microstructures ranging from 10 nm to more than 1 µm [
17,
18,
19]. Importantly, there is a concentration-dependent structural transition from PPII toward a parallel β-sheet conformation accompanied by the formation of large superstructures that activate macrophages in vitro via Toll-like receptor 4 (TLR4) and TLR2 [
20,
21]. The mentioned secondary structures are essential in protein-protein interaction and might function as a signaling component. The high percentage of Q makes this peptide amphiphilic favoring the formation of hydrogen bonds [
22], a key factor in the self-assembling process, stressing the importance between sequence and morphology [
17].
The second most studied gliadin peptide is the toxic p-31-43, a 13-mer peptide comprising the amino acids 31 to 43 (FPGQQQPFPPQQP). Although the p31-43 peptide is not presented by the HLA-DQ2 [
23], the reason for its toxicity in CeD remains unknown. The mechanism by which p31-43 induces an immune response in celiac patients has been recently attributed to its effects on the endocytic compartment affecting several cell functions such as proliferation, cell motility, and innate immunity activation [
23]. Nanayakkara et al. [
24] recently proposed that p31-43 induces the IFN-α-mediated innate immune response in the CaCo-2 enterocyte cell-line by activating the TLR7 signaling pathway mimicking the immune response triggered by viruses. Recently, it was reported that p31-43 has a PPII secondary structure and can, as well as 33-mer, self-assemble under physiologically relevant conditions [
25]. Even more, p31-43 oligomers have been proposed to be responsible for activating the inflammasome in murine models [
26].
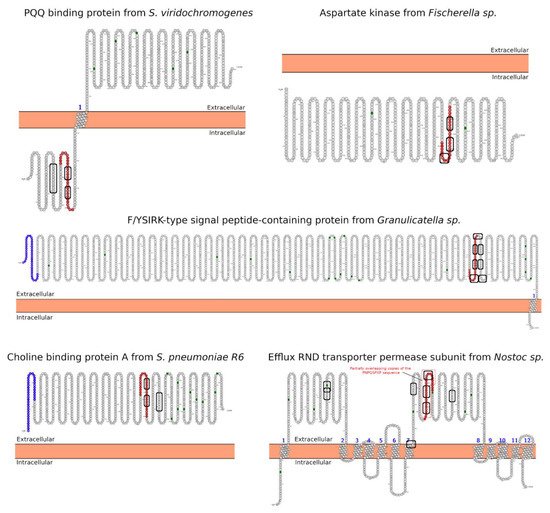
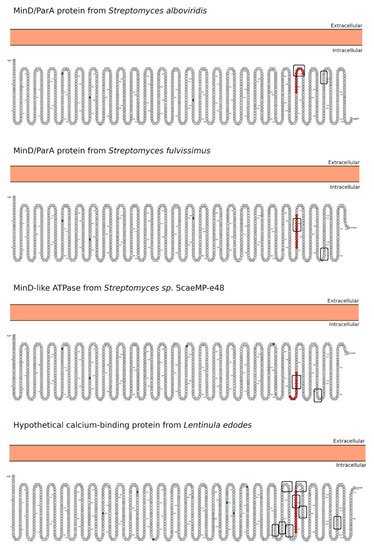
In-depth sequence and structural analysis of foreign proteins sharing high sequence similarity regions with the 33-mer and p31-43 sequences found some interesting novel bacteria connections. The top-five BLASTp hits using the 33-mer were S. viridochromogenes, Fischerella sp., Granulicatella sp., S. pneumoniae and Nostoc sp. Figure 1. In the case of the p31-43 sequence, Streptomyces sp. and L. edodes were found Figure 2. Overall the identified similarity regions reached up to 68% sequence identity in the 33-mer and up to 85% for the p31-43 similarity regions. Importantly, 5 out of 10 identified proteins belonging to the host organisms known to be pathogenic for humans to different degrees.
2. Primary Structure Analysis and Potential Functions
2.1. CeD-T-Cell Epitopes
The molecular mimicry hypothesis considers that pathogens and/or pathogen-derived proteins/peptides sharing molecular and structural similarities with gluten fragments trigger a strong humoral immunological response. After the primary disease is treated, the host immune system recognizes gluten pathogenic fragments as if they are signatures of the bacterial pathogens [
65,
66].
Molecular mimicry is nowadays recognized as a pathogenic mechanism by which infectious or chemical agents may induce autoimmunity and occurs when foreign and self-peptides share similarities at the sequence or structural level, favoring the activation of autoreactive T or B cells in predisposed individuals [
67]. There is extensive documentation of autoimmune diseases that have been associated by molecular mimicry with foreign pathogens such as acute gastroenteritis by rotavirus [
68], autoimmune thyroid diseases by
Helicobacter pylori and Hepatitis C virus [
69], systemic lupus erythematosus by
Leishmania sp. [
70], acute rheumatic fever by group A
streptococci [
71,
72] and recently the SARS-CoV-2 coronavirus, the cause of the worldwide COVID-19 pandemic disease, with the Guillain-Barré syndrome [
73].
In this context, the 33-mer sequence is responsible for the adaptive immune response in CeD because it contains six partially overlapping copies of canonical T-cell epitopes [
14]: three copies of the DQ2.5-glia-α1- (PF/YPQPQLPY) and the DQ2.5-glia-α2 (PQPQLPYPQ) epitope [
74].
2.2. SH3/WW Domains Binders
Although the functions of the similarity regions of the pathogen proteins are unknown, their localization and polyproline II (PPII) propensity (due to the high content of proline and glutamine amino acids) suggest that they could be potential targets in protein-protein interaction with SH3 domains. Many proteins of the Src kinases family carry small modules named Src Homology 3 (SH3) domains with a characteristic β-barrel fold, which usually enables the binding to proline-rich sequences with a PPII conformation [
79]. The SH3 domains are found ubiquitously in all eukaryotes, some prokaryotes, and even viruses [
79]. With more than 300 SH3 domains encoded in the human genome, these are crucial elements in protein-protein interactions in several signal transduction pathways [
80].
In addition, the WW domain mediates specific protein-protein interactions with short proline-rich or proline-containing motifs [
81]. The SH3- and WW domains usually bind to PxxP and xPPx motifs, respectively. In this regard, both the 33-mer and p31-43 peptides could be SH3-binding partners due to their PQLP and PQQP sequences, respectively. Meanwhile, PQSP and PRSP (
Fischerella sp.) and PQAP (
S. viridochromogenes) would be located intracellular (
Figure 1). On the other hand, only the p31-43 and its similar pathogen sequences have the Y/FPPQ sequence predicted to be in the cytoplasm and with the potential capability to bind the WW domain (
Figure 2) [
83,
84]. Thus, both sequences found in gliadin and the pathogen-related proteins may potentially bind to a plethora of proteins in vivo, which could lead to several unknown metabolic (dis-)functions.
Figure 1. Predicted subcellular localization for the BLASTp hits proteins using the 33-mer sequence as query. The localization of the high similarity sequence regions from the five best BLASTp hits is shown in red. The SH3-binding motifs are highlighted with a rectangle. The analysis was performed using the Protter server [
82]. The partially overlapping copies of the PNPQSPXP sequence in the RND efflux pump protein are shown as a red rectangle. The signal peptides are shown in blue and N-glycosylation motifs are marked as green squares.
Figure 2. Predicted subcellular localization for the BLASTp hits proteins using the p31-43 sequence as a query. The localization of the high similarity sequence regions from the four best BLASTp hits is shown in red. The Y/FPPQ-binding motif recognized by the WW domain is highlighted with a rectangle. The analysis was performed using the Protter server [
82]. The N-glycosylation motifs are marked as green squares.
3. Morphology Mimicry
The significance of the term pathogen as “
things” capable of causing human diseases was historically associated with microorganisms. Nevertheless, Griffith’s proposed the pathogenic role of Prions proteins in scrapie in 1967, and the seminal work of Prusiner and coworkers in 1982 laid the groundwork to reconsidering the meaning of the term. Nowadays, the term pathogen was replaced by the widely accepted
infectious agent, which includes biomolecules such as proteins. However, as suggested by Methot and Alizon, a more complex scenario needs to be considered as an ecological, evolutionary, and immunological context takes a prominent role in the host-pathogen interaction [
85].
In this broad scenario, it could also happen that gluten peptides that share structural/morphological similarities with pathogens have latent pathogenicity and, although initially innocuous to the host, after their accumulation and oligomerization with the conformational transition toward amyloid structures, start to be recognized by the host innate immune as non-replicating pathogens. Interestingly, all the identified host pathogens in the BLASTp search share a rod-like morphology that is linearly organized. It is very similar to the morphology of the 33-mer superstructures and, to some extent, to p31-43 oligomers.
Several studies in vitro and in vivo agreed that particles in the size range of 40 nm to 10 μm are the most immunologically active [
86,
87,
88], which fits well with the range of the 33-mer oligomers (10 nm to more than 1 µm). Paul et al. demonstrated that target shape plays a more prominent role than size in the phagocytosis process [
89]. The rod-like morphology of gliadin peptides and their capability to form larger superstructures may be sufficient to generate an early immune response and might serve as general disease-causing signals.
As shown previously in macrophages, only the presence of the larger 33-mer structures activates TLR4 and TLR2 [
21]. Additionally, the p31-43 oligomers are involved in the inflammasome activation in murine models [
26]. Under cumulative conditions due to their inadequate proteolysis, the morphology of the peptide oligomers and particularly the formation of 2D and 3D nano- and microstructures could trigger an early innate response. Unluckily, the presence of canonical T-cell epitopes in the 33-mer sequence triggers the adaptive immune response in CeD patients.
Additionally, it is reported that gliadin acts as a modulator of human microbiota [
27], and it can not be discarded that changes in the microbiota due to the presence of gluten participate in GRDs [
28,
29]. The gliadin superstructures’ presence could also interfere negatively in the initial attachment and subsequent colonization of beneficial bacteria. Another possibility could be that the morphological similarities with pathogenic bacteria favor their attachment and colonization in the mucosa. Notably, both scenarios would lead to dysbiosis, an imbalance of the normal microbiota of the gut [
94].
Pathological overlaps between the protein and 33-mer peptide, e.g., induction of an immune response, were found for the non-covalent extracellular cbpA from S. pneumoniae, which function is to increase the expression of ICAM-1, an early inflammatory maker. In this regard, a specific variant of ICAM-1 with an arginine at position 241 is a predisposing factor for the development of CeD in adulthood. Another significant finding is Granulicatella sp., which is found in the gut and reported in the case of CeD, and the corresponding pathogen-related protein has potential celiac disease T-cell recognition motifs. The molecular and structural similarities with Granulicatella sp. point out the necessity to investigate the role of these pathogens in the development of CeD by molecular mimicry mechanisms.
In summary, these findings stress the importance of further experimental research in the field of gut microbiota, particularly with their connection to CeD and other GRDs, with the final aim to improve the health and life quality of gluten-susceptible people.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22179278