2. Risultati e discussione
In questa voce vengono confrontate due diverse tecniche di estrazione per l'estrazione di PAE da bevande calde generalmente consumate in contenitori di plastica. I PAE possono migrare dalle pareti del contenitore nel contenuto di esso, poiché non sono legati chimicamente al polimero plastico [ 18 ]. Poiché studi recenti hanno dimostrato i loro possibili effetti negativi sull'uomo [ 29 , 30 ], è necessario un metodo di estrazione sensibile e affidabile per la loro estrazione e la conseguente quantificazione nelle bevande più consumate.
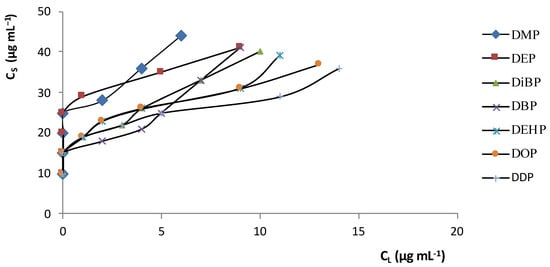
Al fine di creare le migliori condizioni analitiche per SPE, sono state studiate le isoterme di adsorbimento e le curve di sfondamento. La Figura 2 mostra le isoterme di distribuzione per i PAE tra la fase in soluzione acquosa e l' adsorbente C 18 (C S è la concentrazione di soluto in fase solida e C L la concentrazione di soluto in fase liquida). Le curve sono spostate verso la fase solida (C S >> C L ,) a diverse concentrazioni di PAE (basse e alte); tale presenza significa che i composti (soluti) hanno un'affinità verso l'adsorbente.
Figura 2. Isoterme di distribuzione delle PAE tra l' adsorbente C 18 e la soluzione acquosa a 25 °C.
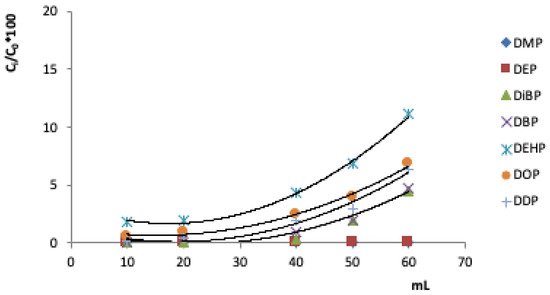
Un altro parametro importante nella valutazione della metodologia SPE riguarda le curve di sfondamento. Consentono infatti di determinare il volume massimo che attraversa la cartuccia senza perdita di analita. Al di sopra di tale volume la fase adsorbente non è in grado di trattenere gli analiti, ed inizia a rilasciare analiti nell'eluato. La Figura 3 mostra le curve di svolta per i sette PAE studiati, ovvero la frazione dell'analita (Ci/C 0 %) rispetto al volume (mL).
Figura 3. Curve di svolta dei PAE studiati in 100 mL di soluzione addizionati con 40 µg mL -1 di ciascun analita sulla cartuccia C 18 (100 mg).
Le curve di sfondamento ci permettono di trovare il volume di sfondamento, cioè il volume massimo per trattenere quantitativamente i composti studiati da parte dell'adsorbente. Il volume di passaggio teorico era di 40 ml, utilizzato anche come volume di passaggio sperimentale.
Tabella 1 mostra i recuperi PAE per ciascun solvente testata in questo studio, cioè acetone, acetato di etile, n eptano, toluene, iso ottano, n pentano, etanolo e cloroformio. Acetone, acetato di etile e n- eptano sono stati in grado di estrarre PAE, con prestazioni diverse (tra 40,5% e 100,2%, 21,8% e 101,9% e 61,4% e 94,7%, rispettivamente). Gli altri solventi quali toluene, iso ottano, n pentano, etanolo e cloroformio mostrano molto bassi recuperi. Guardando i dati ottenuti, il miglior solvente di estrazione è stato l' n- eptano.
Tabella 1. Recuperi (%) di ogni PAE in relazione ai diversi solventi utilizzati per l'estrazione. Tra parentesi sono riportati i coefficienti di variazione (cv%) 1 .
Per quanto riguarda i parametri analitici, la Tabella 2 mostra i principali risultati ottenuti mediante la tecnica SPE seguita dall'analisi GC-FID applicata ad una matrice reale, ovvero il caffè. In particolare, i sette PAE sono stati studiati nell'intervallo di linearità 5-500 µg mL -1 (eccetto DEP e DDP, indagati nell'intervallo 10-500 µg mL -1 ), mostrando R 2 > 0,9938 e ottenendo percentuali di recupero comprese tra 75 e 95%, ad eccezione di DMP (0,9862 e 43,2%, rispettivamente).
Tabella 2. I coefficienti di correlazione (R 2 ) nell'intervallo 5-500 mg mL -1 , limite di rilevamento (LOD), limite di quantificazione (LOQ), i recuperi e intra-giorno e precisione inter-giorno insieme con deviazione standard relativa ( RSD) in campioni di caffè di ciascuna PAE indagata mediante approccio SPE.
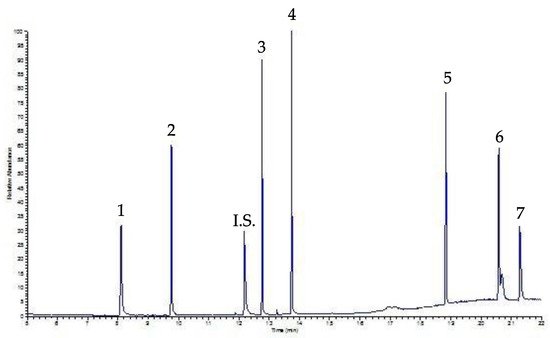
Al fine di validare il protocollo analitico, è stata studiata la sensibilità del metodo in termini di limite di rilevabilità (LOD) e limite di quantificazione (LOQ). LOD e LOQ sono stati determinati sulla base dei criteri proposti dalla International Conference on Harmonization, ovvero Validation of Analytical Procedure: un analita che produce un picco cromatografico pari a tre volte, LOD, o sette volte, LOQ, la deviazione standard del rumore di fondo. Il metodo di estrazione utilizzato ha mostrato il valore di LOD e LOQ compreso tra 0,1 e 0,9 µg mL - 1 e 1,3 e 8,2 µg mL -1, rispettivamente, a conferma della bontà della metodologia analitica. Precisione e accuratezza sono state studiate sulla base della ripetibilità del metodo in un giorno (intragiornaliero 1 e 2) e in tre giorni consecutivi (intragiornaliero). La deviazione standard relativa (RSD) è riportata anche nella Tabella 2 per le misurazioni intraday e interday, comprese rispettivamente tra il 3,9 e l'11% e tra il 5,2 e il 14,7%. Infine, la Figura 4 mostra un cromatogramma di un campione di bevanda, caffè, addizionato con una soluzione di miscelazione contenente 30 µg mL -1 di ciascun PAE dopo la procedura SPE; i picchi sono ben risolti e ordinati.
Figura 4. Cromatogramma SPE-GC-FID di un campione di caffè addizionato con 30 µg mL -1 di ciascun PAE. Picchi: 1 DMP, 2 DEP, IS Internal Standard, 3 DiBP, 4 DBP, 5 DEHP, 6 DOP, 7 DDP; per condizioni sperimentali: vedi testo.
Per quanto riguarda invece DLLME, il protocollo originale di questa tecnica di estrazione si basa su un sistema ternario, che estrae analiti in soluzione acquosa. I solventi disperdenti ed estrattivi vengono aggiunti rapidamente alla soluzione acquosa in modo da ottenere una soluzione torbida. Il solvente disperdente trasporta il solvente di estrazione nella soluzione acquosa, aumentando la superficie di contatto tra gli analiti esaminati e il solvente di estrazione, come mostrato in Figura 2 . In questo lavoro, la dispersione è stata ottenuta mediante vortex e ultrasuoni, come approfondito in un precedente articolo [ 28]. In questo caso abbiamo eliminato l'aggiunta di NaCl perché si è notato che la rottura della microemulsione è assicurata solo dalla fase di centrifugazione, consentendo una riduzione delle fasi del metodo e, quindi, dei tempi di analisi.
Utilizzando UVA-DLLME come tecnica di estrazione, gli autori sono stati in grado di ottenere percentuali di recupero comprese tra il 70,2 e il 104,0%, riportate nella Tabella 3 . Inoltre, LOD e LOQ ottenuti con questo approccio sono leggermente migliori di quelli riportati nella tabella 2 e nella precisione infragiornaliera e intergiornaliera .
Tabella 3. Coefficienti di correlazione (R 2 ) nell'intervallo 5–500 µg mL -1 , limite di rilevabilità (LOD), limite di quantificazione (LOQ), recuperi e precisione infragiornaliera e intergiornaliera insieme alla deviazione standard relativa ( RSD) in campioni di caffè di ciascuna PAE indagata mediante approccio UVA-DLLME.
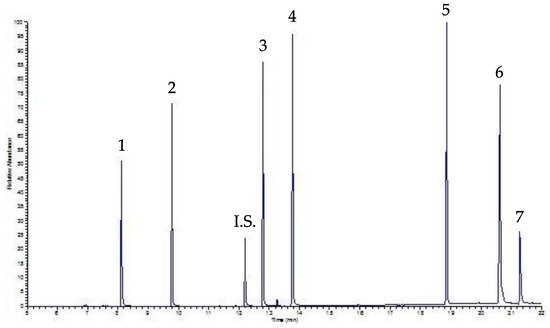
Infine, la Figura 5 mostra il cromatogramma dello stesso campione analizzato in Figura 4 (ovvero un campione di bevanda, precisamente caffè, addizionato con una soluzione di miscelazione contenente 30 µg mL -1 di ciascun PAE), questa volta elaborato da UVA-DLLME. Come si può vedere, il cromatogramma è ancora ben risolto e chiaro, ma in questo caso la linea di base è migliore, consentendo un LOQ inferiore a quello determinato dall'approccio SPE.
Figura 5. Cromatogramma UVA-DLLME-GC-FID di un campione di caffè addizionato con 30 µg mL -1 di ogni PAE. Picchi: vedere la Figura 4 ; per condizioni sperimentali: vedi testo.
In generale, sia le tecniche SPE che UVA-DLLME hanno mostrato recuperi percentuali buoni e soddisfacenti. La tecnica UVA-DLLME ha mostrato recuperi più elevati rispetto alla SPE, compresi rispettivamente tra il 70,2 e il 104,0% e il 77,7 e il 93,6%. I LOD e LOQ ottenuti sono sufficienti per determinare e quantificare i PAE nelle bevande calde. In particolare, l'UVA-DLLME ha mostrato valori di LOD e LOQ inferiori rispetto a quelli di SPE, il che evidenzia la maggiore sensibilità dell'SPE. Inoltre, la tecnica SPE ha il vantaggio di ottenere fattori di pre-concentrazione maggiori, rispetto alla UVA-DLLME, mentre quest'ultima tecnica riduce i tempi di analisi per ottenere la soluzione finale da iniettare nel GC, oltre ad utilizzare un volume minore di soluzione. I parametri analitici mostrano che la tecnica UVA-DLLME aveva una maggiore precisione, ottenendo recuperi compresi tra il 70,2 e il 104,0%, con un RSD inferiore al 9,2%. Questi dati confermano la robustezza e la bontà del protocollo analitico. Sebbene la tecnica SPE abbia mostrato un fattore di pre-concentrazione più elevato e una maggiore sensibilità, l'UVA-DLLME ha raggiunto recuperi quantitativi più elevati, con maggiore precisione. Inoltre, UVA-DLLME ci ha permesso di estrarre gli analiti mediante una piccola quantità din- eptano (50 µl).
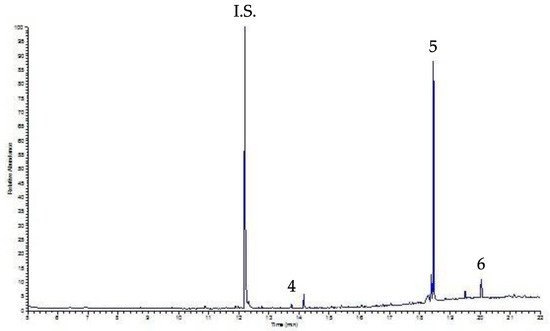
Infine, la procedura UVA-DLLME è stata applicata a campioni di caffè e caffè decaffeinato. La figura 6 mostra il relativo cromatogramma del campione decaffeinato; per entrambi i campioni gli unici picchi rilevati sono stati DEHP e DOP (DBP è inferiore al LOQ da valutare), a livelli non influenti sulla salute umana.
Figura 6. Cromatogramma UVA-DLLME-GC-FID di un campione di caffè decaffeinato. Picchi: vedere la Figura 4 ; per condizioni sperimentali: vedi testo.
Anche se questi dati non mostrano una situazione preoccupante, il prossimo passo di questo approccio è confermare i risultati ottenuti dall'analisi gascromatografica di spettrometria di massa (GC-MS); questo metodo di analisi può raggiungere LOQ molto bassi ed evitare artefatti positivi/negativi che potrebbero influenzare i risultati.