Mitochondria are clustered around the replication sites of several viruses and decrease the supply routes for energy and metabolites, resulting in increased viral progeny viruses. In a viral infection, viruses generate cellular stress, which causes mitochondrial redistribution.
1. Introduction
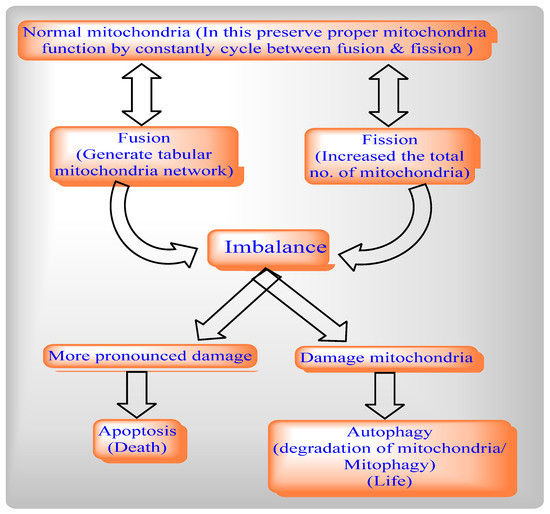
The mitochondrial dynamics network involves two cycles, mitochondrial fission and Mitochondrial Fusion, to help maintain the functional capacity of mitochondria by distribution of mitochondrial contents, energy conductance, and responsiveness to cellular cues. Thus, mitochondrial dynamics govern their communication and interaction with other cellular organelles.
By balancing between two opposite processes, mitochondrial fission and fusion, mammalian cells maintain the overall shapes of their mitochondria. The Fis1 protein has a TM domain with the help of the C-terminal of mitochondria anchored into the mitochondrial outer membrane [3]. Drp1 does not prevent localized mitochondria via the knockdown of Fis1 with RNA interference [7]. By network lengthening, MFF release the Drp1 foci from the mitochondrial outer membrane, whereas, with the help of mitochondrial fission and the physical interaction between the mitochondrial fission factor (MFF) and Drp1, MFF overexpression stimulates mitochondrial fission [6].
Mitochondrial fission includes Drp1 and Fis1, whereas mitochondrial fusion includes Mfn1, Mfn2, and OPA1. The deletion of the Drp1 gene causes mitochondrial enlargement, the increased opening of the mitochondrial permeability transition pore (MPTP), apoptosis, and lethal dilated cardiomyopathy (DCM) [25] by inhibiting mitochondrial fission, whereas deletion of Mfn1 and Mfn2 disrupts mitochondrial structure and respiratory chain function [26]. An imbalance between mitochondrial fusion and fission compromises mitochondrial integrity during aging [27,28,29]. Mitochondrial from aged C. elegans is indicated by a significantly enlarged and swollen ultrastructure, which is accompanied by decreasing O2 consumption, increasing carbonylated proteins and decreasing mitochondrial SOD activity [30].
In healthy mitochondria, PINK1 contains a mitochondrial target sequence (MTS), which translocates to mitochondria and is imported to the IMM by translocase of the outer mitochondrial membrane (OMM) and inner mitochondrial membrane (TIM). Following this, PINK1 is degraded by downstream proteolytic events.
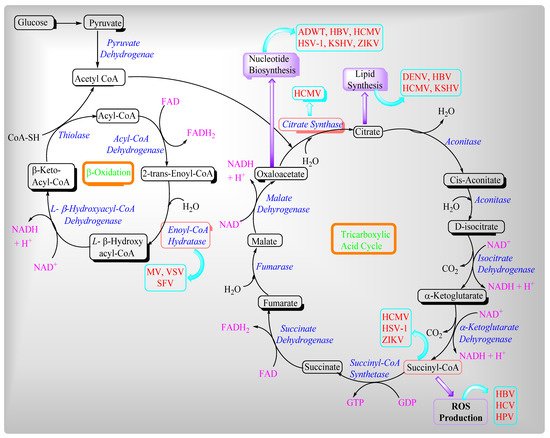
2. Viruses and Their Effects on Mitochondrial Metabolites
In the host cell, viruses use building blocks such as lipids and amino acids for their virion progeny production, whereas energy causes processes such as viral assembly and release [
113,
114,
115]. Moreover, mitochondria have evolved antiviral counter measures. Viruses mainly influence two different mitochondrial metabolic pathways such as the β-oxidation of fatty acids and the Tricarboxylic acid cycle or Krebs Cycle (
Figure 1). Mitochondria are clustered around the replication sites of several viruses and decrease the supply routes for energy and metabolites, resulting in increased viral progeny viruses. In a viral infection, viruses generate cellular stress, which causes mitochondrial redistribution.
Figure 1. Relationships between mitochondrial fission and fusion, apoptosis and mitophagy.
Slow-replicating viruses target the mitochondria by maintaining cellular energy homeostasis to ensure efficient replication and an extended lifecycle, also avoiding programmed cell death. In contrast, fast-replicating viruses easily cope with cellular metabolic dysfunction.
2.1. Regulation of Ca2+ Homeostasis by Viruses in Host Cells
Involved in various cellular process, Ca
2+ acts as secondary messenger. Among different mechanisms, Ca
2+ can enter through voltage-dependent anion channels [VDAC), also known as mitochondrial porins in outer membrane, into the mitochondrial intermembrane space [
116,
117]. This channel regulates Ca
2+ entry and metabolites based on mitochondrial membrane potential (MMP). Ions such as Na
+, H
+, and Ca
2+ exchange across the mitochondrial membrane resulting in decreased MMP, which depends upon the electron transport chain (ETC). The permeability transition pore (PTP) regulates Ca
2+ efflux via a “flickering” mechanism. In Ca
2+ overload, the PTP are opened for a longer duration which causes the destruction of mitochondrial functions. In the inner-mitochondrial membrane, oxidative stress, Ca
2+ overload, and ATP depletion induce the formation of a non-specific permeability transition pore (PTP), which is also responsible for damage to the MMP. Moreover, viruses regulate MMP in the host cells. The MMP value varies from species to species and organ to organ, based on mitochondrial function, protein composition, and the amount of oxidative phosphorylation activity required in that organ of the body [
118].
At the early stage of virus infection, viruses prevent apoptosis from resulting in the prevention of the host immune response and promote cell replication. On the opposite side, at a later stage of virus infection, viruses induce apoptosis and release the progeny virions for dissemination to the surrounding cells.
2.2. Role of Viruses in Modulating Mitochondrial Antiviral Immunity
Viruses attack cells to generate interferon via activating a variety of signal transduction pathways. Pathogen-associated receptors (PRRs) such as the toll-like receptor (TLRs), nucleotide oligomerization domain (NOD) like receptor [NLRs), and retinoic acid-inducible gene (RIG-I) like receptor (RLRs), recognize the pathogen-associated molecular atoms (PAMPs) of viruses which are present inside the cell. PRRs directly activate immune cells [
119].
Mitochondria are associated with RLRs such as the melanoma differentiation-associated gene 5 (Mda-5) and retinoic acid-inducible gene I [RIG-I), which recognize the dsRNA. RIG-I has two terminuses. The N-terminus contains caspase activation and recruitment domains (CARDs) and includes proteins such as mitochondrial antiviral signaling (MAVS), IFN-β promoter stimulator 1 (IPS-1), virus-induced signaling adaptor (VISA), or the CARD adaptor-inducing IFN-β (CARDIF) protein. On the other hand, the C-terminus includes RNA helicase activity [
120] which binds to unmodified RNA produced by a viral polymerase in an ATPase-dependent manner, resulting in the exposure of its CARD domain and activating a downstream effector which leads to the formation of enhanceosome-triggering [
121] NF-kB production.
Mitochondrial Antiviral Signaling (MAVS) contains a proline-rich region on the N terminal CARD and the hydrophobic transmembrane (TM) on the C-terminal, which targets the protein in the mitochondrial outer membrane [
122]. Thus, it plays an essential role in antiviral defense in the cells. The overexpression of MAVS activates NF-kB and IRF-3, which produce type 1 interferon responses. By interacting MAVS with VDAC [
123] preventing apoptosis and the opening of MPTP, the virus cleaves the MAVS from the mitochondrial outer membrane and reduces interferon response [
124,
125].
For example:
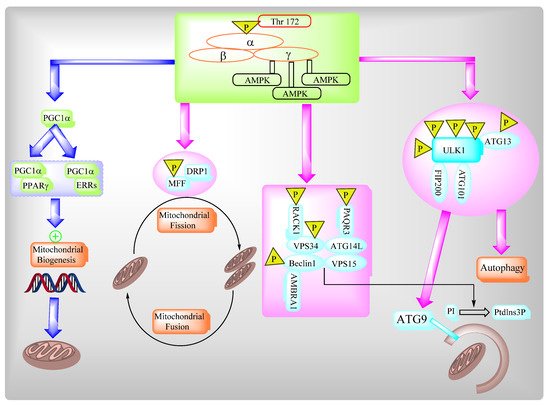
2.3. AMPK Governs Autophagy and Mitochondrial Homeostatsis
The AMP-activated protein kinase (AMPK) complex consists of different subunits including a catalytic α-subunit and two regulatory subunits, β and γ. The AMPK complex senses low cellular ATP levels to increase growth control nodes and the phosphorylation of specific enzymes which produce ATP or lower ATP consumption. AMPK plays an important role in multiple biosynthetic pathways under low cellular energy levels via direct and indirect targeting of the functions of different protein targets of AMPK (Figure 2).
Figure 2. Viruses’ influence on beta-oxidation and the TCA cycle.
2.4. Role of SRV2 in Mitochondrial Dynamics
Ras val-2 (SRV2) is a pro-fission protein that promotes interaction between Drp1 and mitochondria [
131], then oligomerizes Drp1 around mitochondria to form a ring and cut the mitochondria into several fragments. Thus, it has a vital role in mitochondrial shape and fission [
132]. The protein SRV2 also increases the expression of F-actin (as stress fiber) and it provides an adhesive force which helps Drp1 to complete mitochondrial contraction [
133,
134] which facilates mediated mitochondrial fission [
135]. Macro phase stimulating 1 (Mst 1) is a key factor in the Hippo signaling pathway. The loss of Mst 1 maintained mitochondrial homeostasis [
136] by the attenuation of renal ischemia-reperfusion injury as well as in cardiomyocytes, improving mitochondrial performance by autophagy and enhanced cardiomyocyte viability. Additionally, Mst 1 has a role in SRV2-related mitochondrial fission.
2.4.1. SRV2 in Various Functions of Mitochondria
Mitochondrial fission is promoted by the LPS-mediated upregulation of SRV2 [
137,
138]. Loss of SRV2 attenuates mitochondrial fission, protects cardiomyocytes against LPS-induced stress, and improves cell survival and sustained cardiomyocyte function [
139].
SRV2 overexpression promotes mitochondrial fission and leads to cardiomyocyte death and mitochondrial damage [
140]. Thus, the loss of SRV2 exerts an antioxidative effect in cardiomyocytes by inhibiting mitochondrial fission.
With regard to mitochondrial ETC activity, the knockdown of SRV2, LPS, and FCCP have similar effects and decrease ETC transcription. The inhibition of mitochondrial fission prevents the LPS-induced dysregulation of cardiomyocyte energy metabolism [
141,
142,
143].
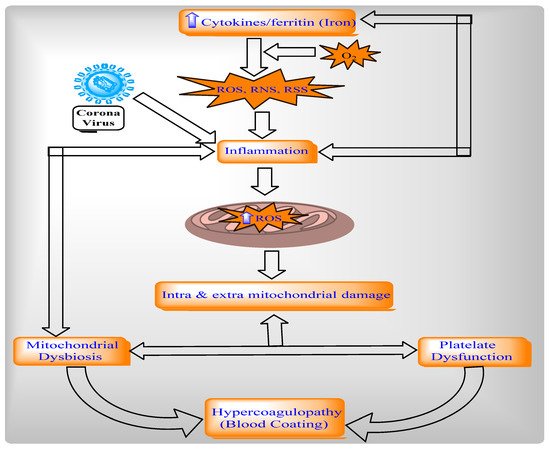
2.4.2. Relationship between Mitochondria, Oxidative Stress, and Inflammation in COVID-19
The protein ROS increases via inflammatory cytokines, such as TNF-alpha in mitochondria, and directly stimulates a generation of pro-inflammatory cytokines [
144]. The ROS in mitochondria is modulated by IL-6 and IL-10. Mitochondrial metabolism is altered through intracellular cascades, which are triggered by inflammatory mediators and immune sentinels. The serum of patients with COVID-19 contains cytokines like TNF- alpha and IL-6, which obstruct mitochondrial oxidative phosphorylation, ATP production, and produce ROS in the cell [
145,
146]. These ROS-altered mitochondrial dynamics permeabilize the mitochondrial membrane and ultimately cause cell death. Additionally, ROS production and mitochondrial content (such as mtDNA) are released into the cytosol and the extracellular environment [
147,
148]. After this, ROS activates NLRP3 inflammasomes and produces pro-inflammatory cytokines such as IL-1beta and induces the production of IL-6 via inflammasome-independent transcriptional regulation [
145,
146,
149,
150]. Thus, ROS contributes to mitochondrial dysfunction (
Figure 3 and
Figure 4). Cytokines can indicate COVID-19 disease severity. Patients with COVID-19 have a large number of pro-inflammatory cytokines (CXCL-8, IL-6, CCL3, CCL4, and IL-12) due to human alveolar epithelial cells with dysfunctional mitochondria [
151]. Thus, these cells impair repair responses and reduce responsiveness to corticosteroid (
Figure 4).
Figure 3. AMPK regulates a variety of metabolic processes.
Figure 4. Mitochondria dysfunction in the pathogenesis of COVID-19.
2.5. Different Pathways to Reposition Common Approved Drugs against COVID-19
The World Health Organization reported that most repositioned drugs modulators, under clinical investigation against COVID-19, act through different pathways such as UPR, autophagy, the NLRP3 inflammasome, and mitochondrial permeability transition pores [MPTP] (Table 1).
Table 1. List of drugs which targeted SARS- CoV related pathways.
| Therapeutic Category |
Mechanism of Action |
| |
Autophagy |
UPR stress |
MPTP |
NLRP3 Inflammasome |
| Activator |
Modulator |
Inhibitor |
Suppressor |
Modulator |
Modulator |
Inhibitor |
| Immunosuppressant |
Rapamycin, Tacrolimus, Everolimus [152] |
|
Cyclosporin A [153,154] |
|
| Anticancer |
Rapamycin, Tersirolimus, Everolimus [152], Gefitinib [155], Temozolomide [155] |
|
Bortezomib, Celecoxib [155] |
Sunitinib [156] |
|
Thalidomide [157] |
| Antidiabetic |
Metformin [152] |
|
Pioglitazone [156], Exenatide, Vildagliptin [158], Berberine [159] |
Liraglutide [159] |
|
Glyburide [157,160] |
| Dietary supplement |
Trehalose, Resveratro l [152] |
|
Curcumin [156] |
Quercetin [161] |
|
| Antipsychotic |
Lithium [152], Fluspirilene, Trifluperazine, Pimozide [162], Bromperidol, Chlorpromazine [163,164], Sertindole, Olanzapine, Fluphenazine, Methotrimeprazine [165], Prochlorperazine [164] |
Clozapine [165] |
|
Haloperidol [166,167,168], Etifoxine [169,170] |
|
| Antiepileptic |
Carbamazepine, Sodium valproate [152] |
|
| Antihypertensive |
Verapamil, Nimodipine, Nitrendipine [152], Nicardipine, Amidarone [162], Rilmenidine, Clonidine [171], Minoxidil [163] |
|
Isoproterenol [156], Valsartan, Lowsartan, Olmesartan, Telmisartan [158], Guanabenz [172], Bisoprolol, Propranolol, Metoprolol [159] |
|
Ifenprodil [166,167,168], Diazoxide, Nicorandil, Tadalafil, Perhaxiline, Carvedilol [153,154] |
|
| Antidiarrheal |
Loperamide [162] |
|
| Ca+ regulator |
Calcifediol [171] |
|
| Anti-infective |
Nitazoxanide [171] |
| Antidepressant |
Nortriptyline [171] |
|
Clomipramine [163] |
Trazodone [173] |
|
| AnticholesteremiC agent |
Simvastatin [170] |
|
Atorvastatin [159] |
|
| Antiemetic |
Chlorpromazine [163,164], Prochlorperazine [164] |
|
Haloperidol [166,167,168] |
Thalidomide [157] |
| Minercorticoid replacement agent |
Fludraocortisone [163,164] |
|
| Antitussive |
Noscapine [163,164] |
|
Carbetapentane, Dextromethorphan [166,167,168] |
|
| Anti-allergic |
Clemastine [163] |
|
| Chelating agent |
Defeiprone [174] |
| Antihelmintic |
Niclosamide [175] |
|
Quimacrine [176,177] |
|
| Skeletal muscle relaxant |
|
Baclofen [178] |
|
| Gastrointestinal |
|
Pantoprazole [155] |
|
| Macrolide antibiotic |
Azithromycin [163] |
| Ocular drug |
Verteporfin [163] |
| Antiprotozoal drug |
Quimacrine [176,177], Chloroquine, Hydroxychloroquine [171] |
| Urea cycle disorder agent |
|
Thenylbutyrate [155,156] |
|
| Hypolipidemic agent |
Pravastatin [156], Fenofibrate [158] |
| Anti-Alzheimer’s |
|
Donepeziol [166,167,168] |
|
| Anti-Parkinsonian |
Pramipexole [179] |
| Neuroprotective agent; anti-ALS drug |
Edaravone [153,180] |
| Anti-arthritic |
|
Anakinra [157] |
| Anti-inflammatory agent |
|
Celecoxib [155] |
|
Anakinra [157], Tranilast [157,181] |
| Anti-insomia agent |
|
Melatonin [182] |
|
3. Expert Opinion
Mitochondria are membrane-bound cell organelles which produce energy in the form of adenosine triphosphate (ATP) as well as regulating various intracellular functions like metabolism, bioenergetics, cell death, innate immune signaling, and cellular homeostasis. Mitochondria are self-governed by mitochondrial dynamics and mitochondria-selective autophagy or mitophagy. During infection, viruses altered mitochondrial dynamics in order to modulate mitochondria-mediated antiviral immune responses via the alteration of mitochondrial events such as autophagy, mitophagy, and cellular metabolism to facilitate their proliferation.
The pro-fission protein of SRV2 activates mitochondrial fission via the loss of MMP, the ROS-overloading suppression antioxidant system, the depletion of cellular ATP, the release of the apoptotic factor, the activation of the caspase family, and NLRP3 inflammasomes. The protein SRV2 also promotes mitochondria-associated cardiomyocyte apoptosis to cause cardiomyocyte death and mitochondrial damage. The World Health Organization reported that most repositioned drugs modulators, under clinical investigation against COVID-19, act through different pathways such as UPR, autophagy, the NLRP3 inflammasome, and mitochondrial permeability transition pores (MPTP) to inhibit SARS-COV2 propagation. Analysis of the functional significance of mitochondrial dynamics and viral pathogenesis will open up new possibilities for the therapeutic design of approaches to combat viral infections and associated diseases.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22158180